


Kazi Shabbir Anwar1, Tanjina Sharifa2*, Trishita Swarna Roy3
1Head (Pediatric ophthalmology) and Director, Bangladesh Eye Hospital, Dhaka
2Assistant Professor, Department of Pediatrics, Institute of Child and Mother Health (ICMH), Dhaka
3Medical officer, Bangladesh Eye Hospital, Dhaka
*Corresponding Author: Tanjina Sharifa, Assistant Professor, Department of Pediatrics, Institute of Child and Mother Health (ICMH), Matuail, Dhaka, Bangladesh.
Abstract
Stickler Syndrome is an autosomal dominant hereditary disorder that affects the connective tissue, mainly collagen. Here, we report a Bangladeshi family who visited OPD of the Bangladesh Eye Hospital, Dhaka with their 6-year-old female child having complaints of low vision due to cataracts along with facial dysmorphism, speech delay, and hearing impairment. She was diagnosed as a case of Stickler Syndrome clinically and later confirmed by genetic study. Eventually, her 2 younger siblings (1 girl and 1 boy) were also detected as having similar problems (both of them were found to harbor a pathogenic heterozygous variant in the COL11A1 gene, as their eldest sister). Low-grade mosaicism for pathogenic COL11A1 mutation with mild SS Phenotypic traits was also reported in their father.
1. Introduction
Stickler Syndrome is a rare genetically inherited progressive arthro- ophthalmopathy, introduced to the medical community by pediatrician Gunnar B. Stickler in 1965. It is a connective tissue disorder that affects the body’s collagen and is characterized by defective and inadequate collagen (predominantly types II, IX and XI) production. Most known mutations that lead to Stickler syndrome are found on the COL2A1, COL11A1 and COL11A2 genes, which code for the collagens found in eyes, joints and bones. It affects 1 in 7,500 to 10,000 newborns across the globe and both sexes are affected. [1] The only known risk factor for Stickler syndrome is a family history of the condition. The child of an affected parent with dominant inheritance has at least a 50% chance of inheriting the pathologic variant. Suppose both parents are heterozygous for an autosomal recessive Stickler syndrome. In that case, there is a 25% chance of having the disease, a 50% chance of being heterozygous and an asymptomatic carrier, and a 25% chance of being unaffected with no pathologic variant. [2]
The condition has been divided into 4 subgroups based on ocular and systemic clinical findings, with each subgroup also corresponding to a specific genetic defect. [3] Types 1 to 4 of Stickler syndrome are classically inherited in an autosomal dominant fashion, though a significant number of cases were sporadic or came with recessive inheritance (Type 5; STL5).
Table 1: Subtypes of Stickler syndrome
|
Types |
Clinical features |
Gene |
|
Type 1 (STL1) |
Membranous vitreous, congenitalmeg alophthalmos, |
COL2A1 |
|
Type 2 (STL2) |
Beaded vitreous, otherwise similar to type 1 |
COL11A1 |
|
Type 3 (STL3) |
No ocular involvement, otherwise similar to type 1 |
COL11A2 |
|
Type 4 (STL4), |
Membranous vitreous, congenital |
COL9A2, |
|
Type 5 (STL5) |
Megalophthalmos; no systemic findings |
COL9A1, COL9A3 |
The condition is commonly associated with distinctive facial appearance, eye abnormalities, hearing loss, and joint problems. Due to allelic heterogeneity, these signs and symptoms vary widely among affected individuals, even among members of the same family. The ophthalmologic manifestations include premature cataracts, glaucoma, vitreous abnormalities, congenital megalophthalmos causing myopia, radial perivascular retinal lattice degeneration, and retinal detachment. In addition to those, systemic findings may include flattened facial appearance (mid-facial underdevelopment), micrognathia, bifid uvulae and cleft palate, hearing loss (conductive and sensorineural, due to middle and inner ear change), and joint problems (mild spondylo-epiphyseal dysplasia, scoliosis, kyphosis and/ or precocious arthritis). [4]
Diagnosis of Stickler syndrome is clinical, many times it is not detected until after the age of 4 due to the difficulty in slit-lamp examination of infants and young children. The earliest ophthalmic findings noted may be the characteristic radial perivascular retinal degeneration visible by indirect ophthalmoscopy. Audiometric and radiographic evaluation may follow the detection of the typical ophthalmic findings. Targeted molecular genetic analysis is done for the benefit of genetic counseling.
Treatment of Stickler syndrome involves both prophylaxis and treatment of its complications, most commonly retinal detachment (by 360° retinal laser or cryotherapy). Corrective lenses, anti- hypertensives for glaucoma, and cataract surgery may help in retaining vision. Management with hearing aids, speech therapy, joint replacement, and special education by a multidisciplinary team of specialists may be beneficial for the other associated problems.
There is no cure for Stickler syndrome. The prognosis is guarded. Difficult breathing or feeding affects prognosis considerably and needs special care. If glaucoma or retinal detachment is left untreated, the child will become blind.
2. Case Reports Of 3 Siblings
Baby X, a 6-year-old girl, 1st issue of non-consanguineous parents with an upper-middle-class background, visited the OPD of the Bangladesh Eye Hospital, Dhaka with her parents and presented with complaints of decreased vision along with speech delay and hearing impairment. She was diagnosed with a cataract in her left eye. However, the characteristic facial dysmorphism (bilateral exophthalmos, flat face, low-set ears, micrognathia) and associated complaints produced some doubt in the ophthalmologist and he arranged for further clinical evaluation. History showed that her birth was term and uncomplicated, and motor development was age- appropriate. She just started schooling and the issue of low vision was detected by her teacher.
Later she was diagnosed as a case of Stickler syndrome and then confirmed by genetic study (Clinical Exome Sequencing, blood sample collected on 19th July 2022, report on 19th August 2022).
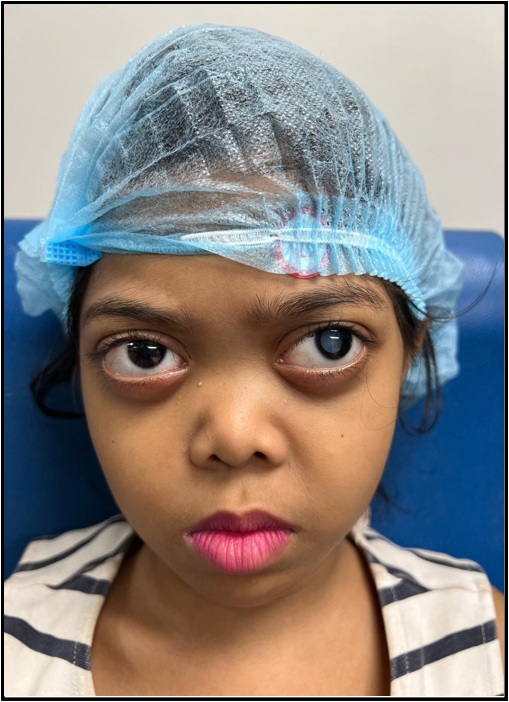
Figure 1: Baby X
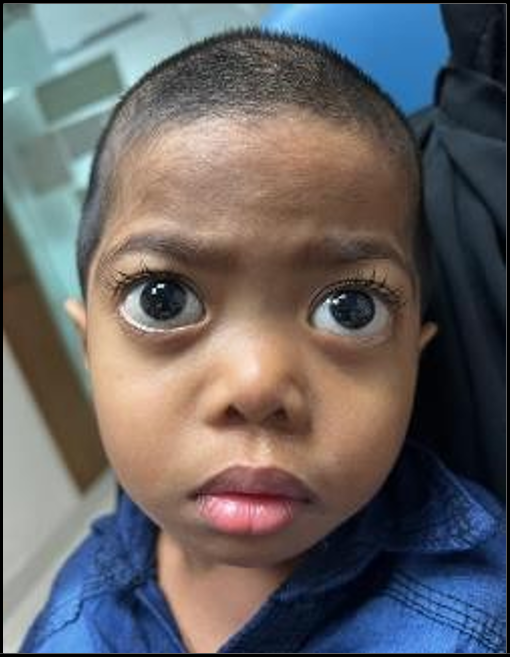
Figure 2: Baby Y
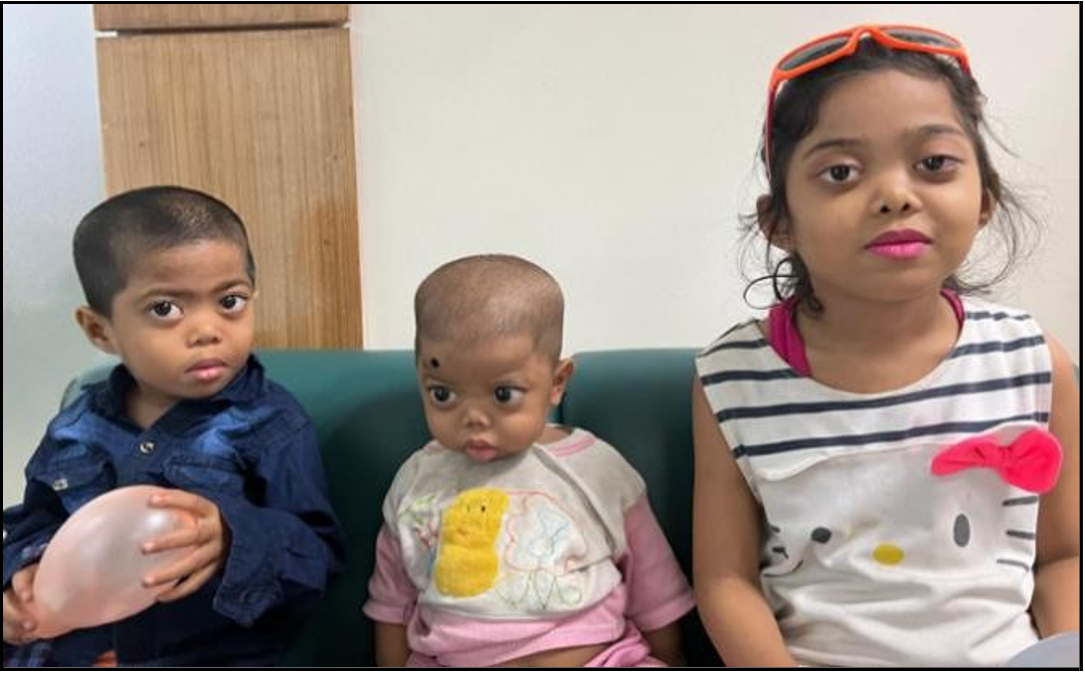
Figure 3: All 3 siblings with Stickler syndrome
So, parental counseling regarding the nature of this disease was done. Then the parents brought their 2nd child ‘Baby Y’ to the OPD of Eye Hospital, a 2-year-old girl, having similar facial dysmorphism as her elder sister, Baby X. She too had delayed speech and doubtful hearing. They requested that Baby Y should also be evaluated for pathogenic variations of the same hereditary disease. The mother was carrying a third child (Fetus Z) at that time. A decision was made to check the pathogenic variants by genetic study for both Baby Y (Clinical Exome Sequencing, blood sample collected on 22nd August 2022) and Fetus Z (Prenatal Sanger variant analysis, Chorionic villus sample collected on 21st August 2022).
Table 2: Summary of the genetic analysis of Baby X and Baby Y
|
|
Gene |
Location |
Variant |
Zygosity |
Inheritance |
Classification |
|
Baby X |
COL11A1(-) |
Intron 50 |
C.3852+1G>A |
Heterozygous |
Autosomal |
Pathogenic |
|
Baby Y |
COL11A1(-) |
Intron 50 |
C.3852+1G>A |
Heterozygous |
Autosomal |
Pathogenic |
Sequence chromatogram and alignment to the reference sequence showed that the variant in intron 50 of the COL11A1 gene [chr1: g,102915630C>T;c.3852+1G>A (5’ splice site)] was detected in heterozygous condition in the Fetus Z with set 1 primer (Baby X) and set 2 primer (Baby Y).
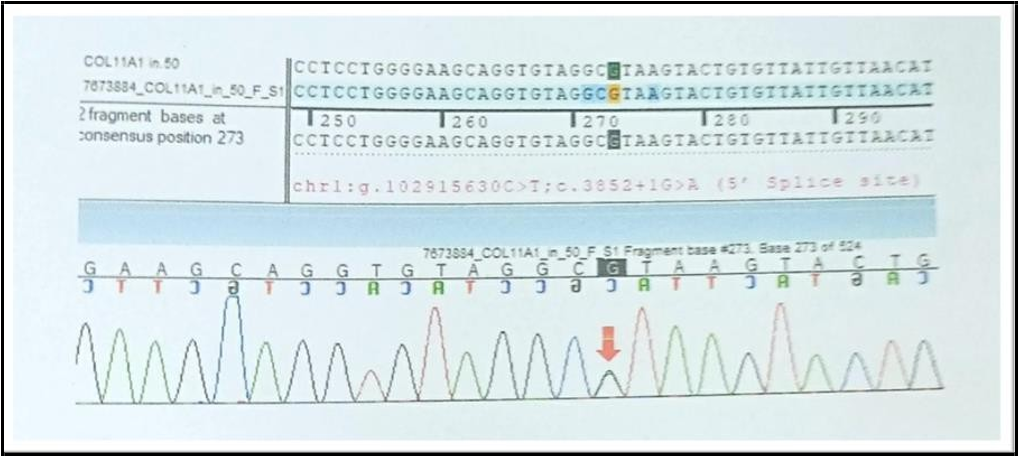
Figure 4: Fetus Z (to be sibling) with set 1 primer
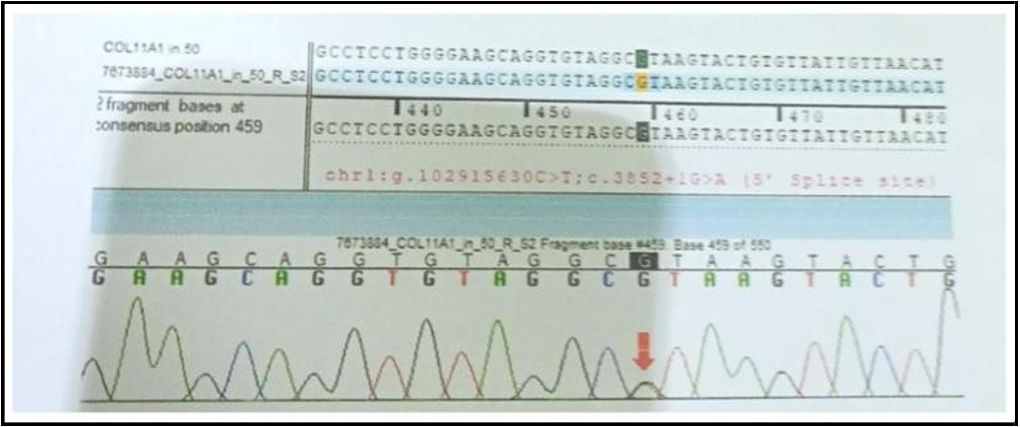
Figure 5: Fetus Z (to be sibling) with set 2 primer
Eventually, the 2 younger siblings (1 girl, Baby Y, and 1 boy, Fetus Z) were also detected as having problems (both of them were found to harbor a pathogenic heterozygous variant in the COL11A1 gene, as their eldest sister). Low-grade mosaicism for a pathogenic COL11A1 mutation with mild SS Phenotypic traits was also reported in their father.
As per Baby X’s condition, Cataract surgery was done on her left eye (I/A with PCIOL under G/A on 26.10.2022) along with other management (Anterior vitrectomy and Peripheral iridectomy). Visual acuity on L/E was 6/9 after the operation. All 3 siblings are now being regularly followed up for any ophthalmological signs/symptoms associated with Stickler syndrome at OPD, Bangladesh Eye Hospital, Dhaka and they are under the care of a multidisciplinary team led by a Neuropediatrician for other manifestations.
3. Discussion
Stickler Syndrome is a multisystem collagenopathy, where all of the genetic mutations are found to have resulted in insufficient and abnormal collagen production. The phenotype analysis revealed ocular involvement and craniofacial dysmorphism as core features of it. Diagnosis of the syndrome is clinical-based, and molecular genetic analysis is used for diagnostic confirmation. To assess the evolution of growth and psychomotor development and to allow an early or timely diagnosis of complications associated with this syndrome, multidisciplinary monitoring is essential. Genetic counseling is important as this syndrome has marked medical and personal consequences, both for the patients and their families.
The purpose of this present study is to report Stickler syndrome in three siblings in a Bangladeshi family, all showing a heterozygous mutation of the COL11A1 gene, identified by Clinical Exome Sequencing. Only ~20% of Stickler syndrome cases are attributed to mutations in the COL11A1 gene. COL11A1 is located on chromosome 1p21, encodes the alpha 1 chain of type XI collagen, and seems to play an important role in fibrillogenesis. [5] Heterozygous pathogenic COL11A1 variants are predominantly splice site alterations and missense variants including glycine substitutions. The splice site alterations usually result in in-frame deletions (exon skipping) and therefore most likely have a dominant-negative effect. Intron 50 (donor splice site) is a mutational hotspot. [6]
It is appropriate to evaluate the older and younger siblings of an index case and molecular genetic testing should be considered as diagnosis, not only assists in treatment and management of the patient but may also help identify other at-risk family members, and in our study, we have just done it. Supportive care and monitoring by multidisciplinary care may lead to a favorable outcome in the form of improved quality of life, maximized function, and reduced complications.
Informed Consent
The written informed consent was obtained from the patient’s legal guardian for the publication of this case, along with pictures.
Declaration Of Interest
The Authors declare that there is no conflict of interest.
References
- Ahmad NN, Dimascio J, Knowlton RG, Tasman WS (1995) Stickler syndrome. A mutation in the nonhelical 3′ end of type II procollagen gene. Arch Ophthalmol. 113(11): 1454-1457.
- Mortier G, Adam MP, Feldman J, Mirzaa GM, Pagon RA, et al. (2000) Stickler Syndrome. Seattle (WA): University of Washington, Seattle.1993–2023.
- Snead MP, McNinch AM, Poulson AV, Bearcroft P, Silverman B, et al. (2011) Stickler syndrome, ocular-only variants and a key diagnostic role for the ophthalmologist. Eye (London). 25(11): 1389-400.
- Carvalho V, Oliveira DR, Ribeiro Silva M, Leite L, Rocha MG, et al. (2022) Stickler syndrome: case report. J Pediatr Neonat Individual Med.11(1): 1-6.
- Brizola E, Gnoli M, Tremosini M, Nucci P, Bargiacchi S, et al. (2020) Variable clinical expression of Stickler Syndrome: A case report of a novel COL11A1 mutation. Mol Genet Genomic Med. 8(9): e1353.
- Geert Mortier (2023) Stickler syndrome. Seattle, WA: University of Washington. 1993–2020.