


Fatimah M. Alabdrabalnabi¹, Khadija M. Alshehabi², Mohammed H. Al Abkary¹, Najib Musaied¹, Deena T. Boqari³, Sumayah Askandarani¹*
¹Multi-Organ Transplant Center, King Fahad Specialist Hospital, Eastern Health Cluster, Saudi Arabia
²Department of Nephrology, Salmaniya Medical Complex, Government Hospitals, Bahrain
³Department of Pathology, King Fahad Specialist Hospital, Eastern Health Cluster, Saudi Arabia
*Corresponding Author: Sumayah Askandarani, Multi-Organ Transplant Center, King Fahad Specialist Hospital, Eastern Health Cluster, Saudi Arabia.
Abstract
Background: MYH9-related disorders (MYH9-RD), such as May–Hegglin anomaly, are rare inherited thrombocytopenias caused by mutations in the MYH9 gene. They are characterized by thrombocytopenia with giant platelets and cytoplasmic inclusions in neutrophils (Döhle- like bodies), often associated with extra-hematologic features including nephropathy, sensorineural hearing loss, cataracts, and elevated liver enzymes. In patients progressing to end-stage kidney disease (ESKD), renal transplantation is the treatment of choice, though bleeding risk, alloimmunization, and infections present significant perioperative and post- transplant challenges.
Case Presentation: We present the case of a 34-year-old Saudi woman with a genetically onfirmed diagnosis of May–Hegglin anomaly (MHA) and a strong family history of thrombocytopenia and end-stage kidney disease (ESKD). She developed proteinuria in early adulthood, which progressed to ESKD, leading to a living unrelated kidney transplant abroad in January 2024. Shortly after the transplant, she experienced hypertensive urgency, severe anemia, thrombocytopenia, and an expanding perigraft hematoma. She subsequently developed Banff 2A acute cellular rejection, which was successfully managed with corticosteroids and antithymocyte globulin. Her postoperative course was further complicated by a ureteric leak and obstruction, requiring nephrostomy placement and ureter-to-ureter anastomosis, as well as recurrent multidrug-resistant urinary tract infections, including Klebsiella pneumoniae, ESBL E. coli, Pseudomonas aeruginosa, and Enterobacter cloacae. Despite these challenges, she ultimately achieved stable graft function, with a serum creatinine of 106 µmol/L at her most recent follow- up in October 2024.
Conclusion: Kidney transplantation offers a feasible therapeutic option for individuals with May–Hegglin anomaly, but it is accompanied by unique and significant challenges. This case highlights the intricate relationship between a predisposition for bleeding, the development of perioperative hematomas, the risks related to alloimmune responses, and the occurrence of recurrent infections. Achieving optimal, long-term outcomes in this population requires an individualized approach, supported by close collaboration within a multidisciplinary medical team.
Keywords: May–Hegglin anomaly, MYH9-related disorders, kidney transplantation, perigraft hematoma, rejection, multidrug-resistant infection
Introduction
MYH9-related disorders (MYH9-RD) are a rare group of autosomal dominant thrombocytopenias caused by mutations in the MYH9 gene, located on chromosome 22, which encodes nonmuscle myosin heavy chain IIA [1]. These conditions are characterized by macrothrombocytopenia and cytoplasmic inclusions in neutrophils (Döhle-like bodies), and may present with extra-hematologic manifestations including nephropathy, sensorineural hearing loss, cataracts, and elevated liver enzymes [2, 3].
Renal involvement is reported in approximately 25–30% of affected individuals, often manifesting as proteinuria and hematuria that progress to focal segmental glomerulosclerosis (FSGS) and end-stage kidney disease (ESKD) before the fourth decade of life [3, 10, 12, 13]. The underlying mechanism involves defective non-muscle myosin heavy chain IIA in podocytes, resulting in cytoskeletal disruption and loss of glomerular filtration barrier integrity [4, 5]. Despite advances in genetic testing, diagnosis is often delayed or misclassified as immune thrombocytopenia, Alport syndrome, or idiopathic FSGS, which may lead to inappropriate treatments [12].
Renal replacement therapy, including transplantation, is indicated for patients with advanced diseases. Kidney transplantation is generally considered successful, as the podocyte defect is intrinsic to the native kidneys and does not recur in the graft [9, 12]. However, patients with MYH9-RD pose unique challenges in the transplant setting: increased perioperative bleeding risk due to macrothrombocytopenia, diagnostic complexity when evaluating graft dysfunction, and heightened susceptibility to sensitization and infections from prior transfusions and urological complications [6, 7, 9].
The majority of reports in the literature are limited to single-patient cases or small series. While these consistently highlight the perioperative bleeding risks in MYH9-related disorders, they seldom provide comprehensive descriptions of vascular complications, rejection or the recurrence of multidrug-resistant infections [7-9, 11]. In this context, we describe the case of a Saudi woman with May– Hegglin anomaly and end-stage kidney disease who received a living unrelated kidney transplant abroad. Her post-transplant period was notable for acute rejection, development of perigraft hematoma, vascular turbulence, ureteric obstruction, and repeated multidrug- resistant infections. Despite these complex challenges, she ultimately achieved stable, long-term graft function.
Case Presentation
A 34-year-old woman from the southern region of Saudi Arabia was diagnosed in childhood with May–Hegglin anomaly (MHA), confirmed by genetic testing following persistent macrothrombocytopenia. Her hematologic profile has been characterized by chronic thrombocytopenia with giant platelets, and she has exhibited a lifelong tendency toward easy bruising, though notably without deep hematomas or visceral bleeding. Her history was negative for other extra-hematological manifestations commonly associated with MHA, such as cataracts or sensorineural hearing loss. She has a strong family history of similar hematological abnormalities, affecting her mother, four maternal uncles, one cousin, and all her siblings, suggesting an autosomal dominant inheritance pattern. Notably, her mother also has end-stage kidney disease (ESKD).
Over the years, the patient required multiple transfusions of blood products, including packed red blood cells and platelets, particularly in the peripartum period following four cesarean deliveries. Despite her bleeding diathesis, she tolerated these procedures without major hemorrhagic complications.
At age 21, during the first trimester of her first pregnancy, she was incidentally found to have proteinuria, which persisted postpartum. Although kidney biopsy was recommended on several occasions to elucidate the underlying etiology, the patient declined. Over time, her proteinuria progressed, and she developed chronic kidney disease (CKD). She initiated maintenance dialysis in November 2021.
In November 2022, she was referred to our center for pre-transplant evaluation. Immunologic workup revealed that she is a highly sensitized candidate, likely due to prior pregnancies and multiple transfusions. Donor-specific antibodies (DSAs) were detected, including moderate anti-DQB1*06:03 (mean fluorescence intensity [MFI] 2700), and weak antibodies against HLA-B*58 and HLA- B*03:01, which may pose challenges in donor matching and increase the risk of antibody-mediated rejection.
The patient subsequently lost follow-up, but returned to our hospital on January 20, 2024, after undergoing a living unrelated kidney transplant abroad via transplant tourism earlier that month. According to a brief external medical report, basiliximab was used for induction, followed by tacrolimus, mycophenolate mofetil, and prednisolone for maintenance. She was discharged on postoperative day five with stable graft function, although no surgical details or perioperative complications were documented.
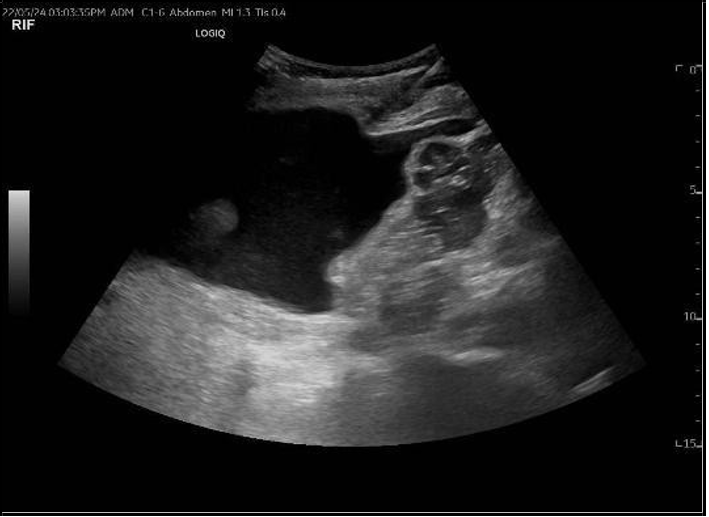
Upon presentation to our emergency department, her vital signs were notable for hypertension (BP 216/118 mmHg) and tachycardia (HR 109 bpm), though she remained afebrile and maintained oxygen saturation of 100%. Physical examination revealed ecchymosis over the right lower quadrant at the surgical site. Laboratory investigations showed severe anemia (Hb 7 g/dL), thrombocytopenia (66 × 10⁹/L), and serum creatinine (68 µmol/L). Abdominal ultrasound demonstrated preserved perfusion of the graft with patent main renal vessels. However, there was a significant increase in resistive index (RI) values within the intrarenal arterial branches, with areas showing a monophasic wave pattern, suggestive of impaired intrarenal flow and warranting close follow-up [Figure 1]. Additionally, elevated velocity at the main renal artery anastomosis site was noted, likely reflecting postoperative changes. A peri-graft fluid collection was identified, with the largest pocket measuring approximately 74 mL, requiring ongoing monitoring for potential complications.
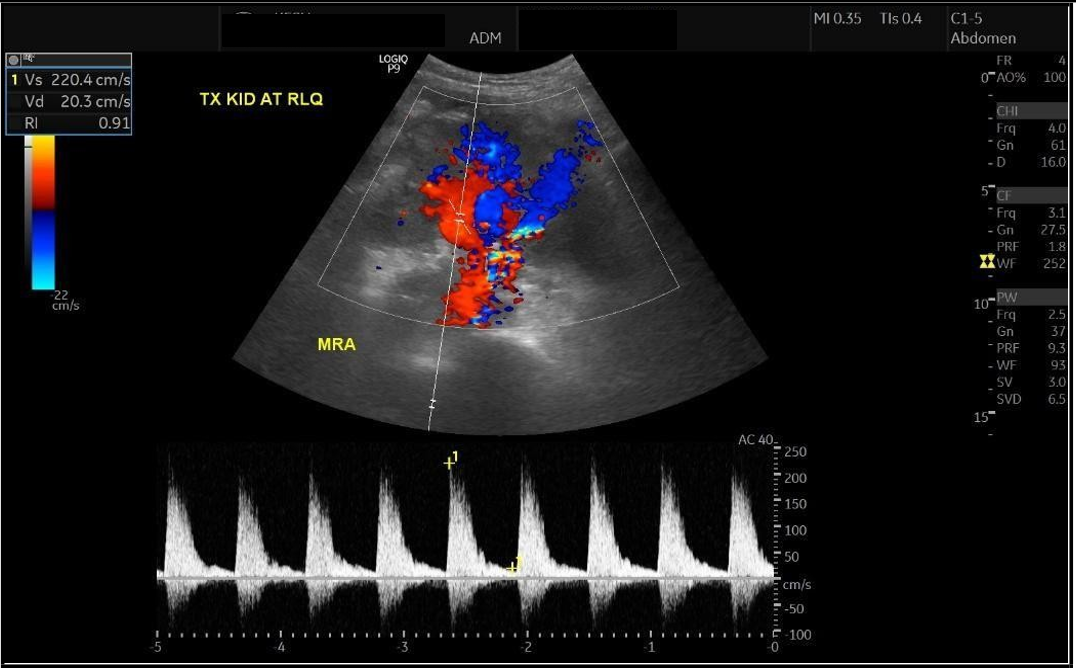 Figure 1: Doppler showing elevated intrarenal resistive index with monophasic waveforms, suggesting impaired intrarenal flow (RI = 0.91).
Figure 1: Doppler showing elevated intrarenal resistive index with monophasic waveforms, suggesting impaired intrarenal flow (RI = 0.91).
Then, the patient was admitted due to concerns about laboratory trends and evolving clinical findings. Repeat labs confirmed a further drop in hemoglobin to 5.4 g/dL, persistent thrombocytopenia (75 × 10⁹/L), and worsening renal function with serum creatinine of 139 µmol/L. These findings, in the context of her hematologic disorder and recent transplant, raised concerns about possible bleeding, graft compromise, or early rejection.
Follow-up graft ultrasound revealed a marked increase in peri-graft fluid collection, expanding from 74 mL to 267 mL, suggestive of a postoperative hematoma or lymphocele [Figure 2]. Following surgical team consultation, the patient was admitted for blood pressure control, red blood cell transfusion, and close monitoring of renal function. After receiving four units of packed red blood cells, her hemoglobin stabilized at 7.4 g/dL, but creatinine continued to rise, reaching 155 µmol/L, with a urine albumin-to-creatinine ratio (ACR) of 762 mg/g.
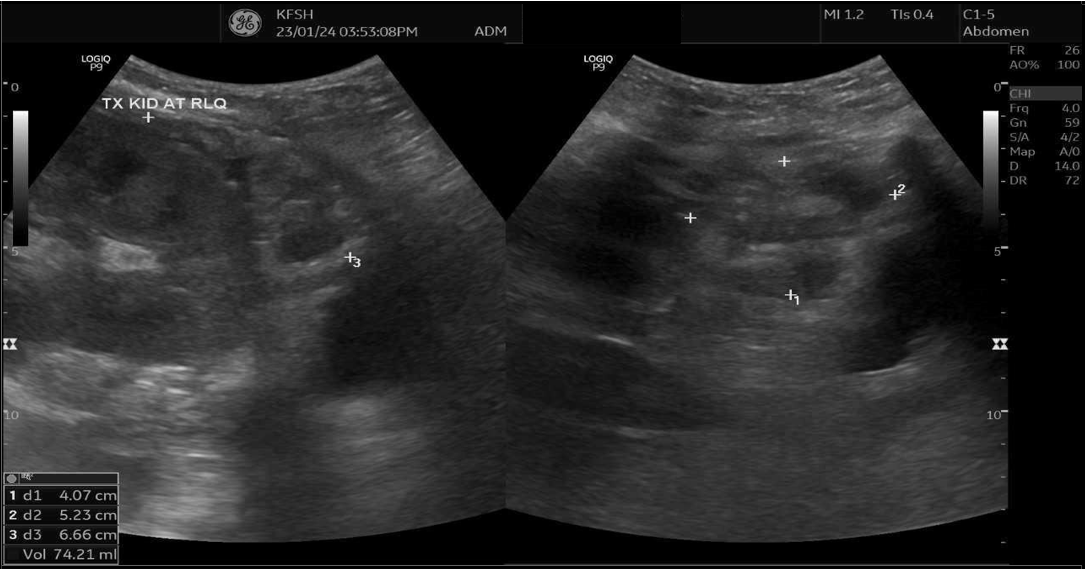 (A)
(A)
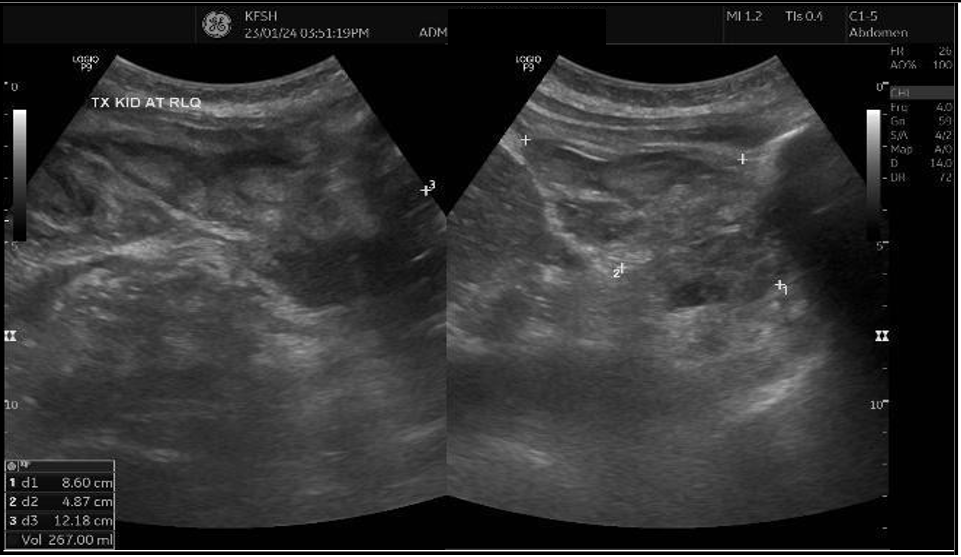 (B)
(B)
Figure 2: (A) Ultrasound Doppler of renal transplant showing perigraft heterogeneous fluid collection measuring 4.0 × 5.2 × 6.6 cm (74 mL) (B) Left lower quadrant collection measuring 8.6 × 4.8 × 12 cm (267 mL).
A percutaneous transplant kidney biopsy performed on January 30, 2024, revealed acute cellular rejection, classified as Banff 2019 vascular type, grade 2A, along with mild acute tubular injury [Figure 3]. The patient was treated with pulse corticosteroids and antithymocyte globulin (thymoglobulin) at a cumulative dose of 3 mg/kg. Her renal function improved significantly, with creatinine decreasing to 56 µmol/L, and she was discharged home on February 4, 2024.
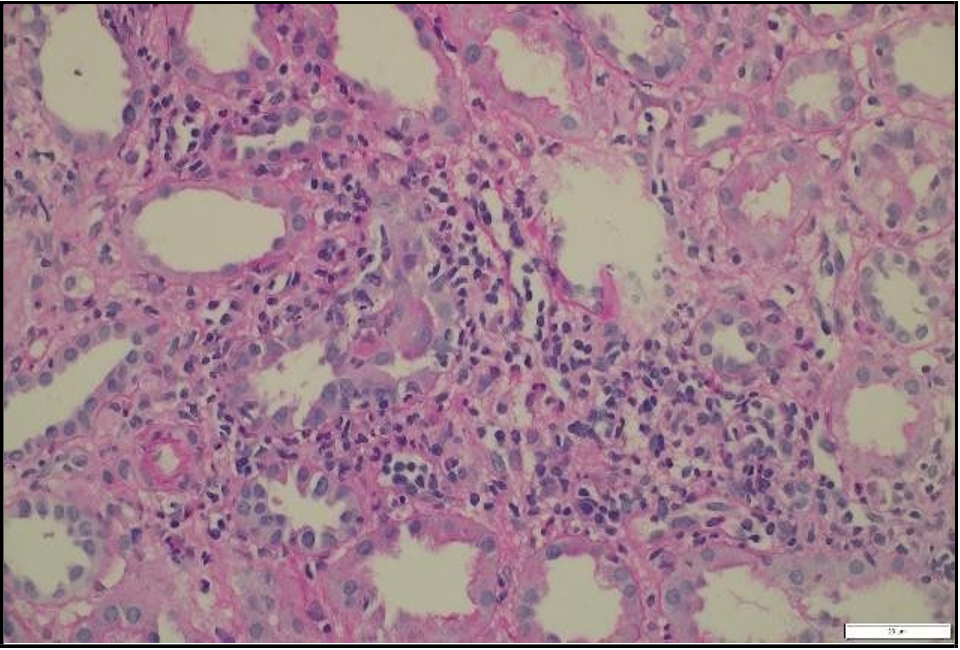
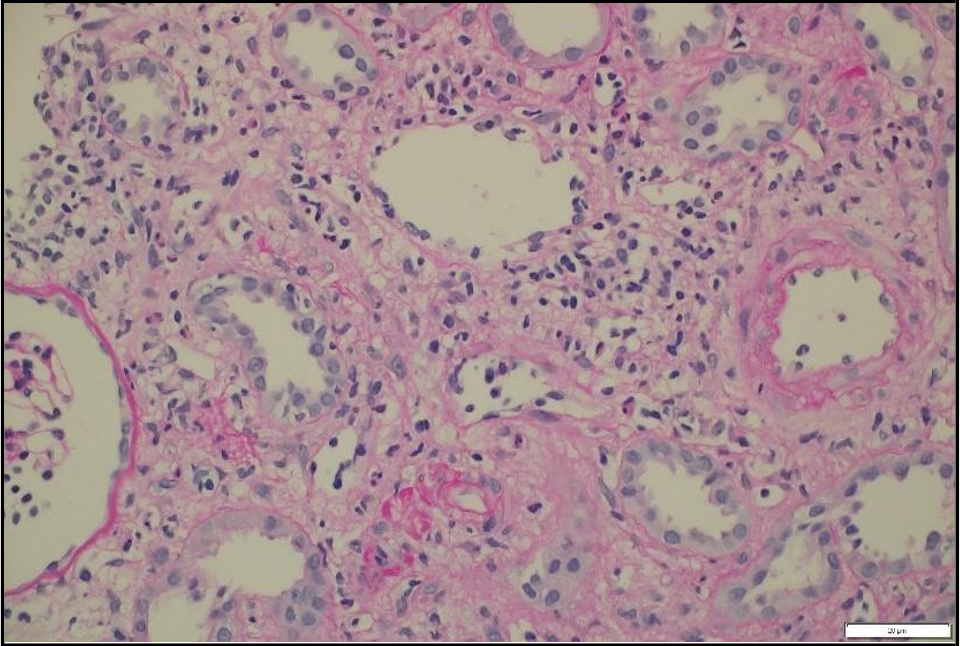 Figure 3: Light microscopy of transplant kidney biopsy showing severe lymphocytic tubulitis (t3), moderate interstitial inflammation (ti2), and mild intimal arteritis (v1), consistent with acute cellular rejection (Banff 201G; vascular type, grade 2A).
Figure 3: Light microscopy of transplant kidney biopsy showing severe lymphocytic tubulitis (t3), moderate interstitial inflammation (ti2), and mild intimal arteritis (v1), consistent with acute cellular rejection (Banff 201G; vascular type, grade 2A).
At discharge and during follow up, her average hemoglobin ranged between 8–10 g/dL, and platelet counts fluctuated between 50–100 × 10⁹/L, consistent with her underlying hematologic disorder. Despite receiving thymoglobulin, she tolerated full immunosuppressive therapy and antimicrobial prophylaxis, including valganciclovir, trimethoprim-sulfamethoxazole (Bactrim), and isoniazid (INH), without hematological complications [Figure 4].
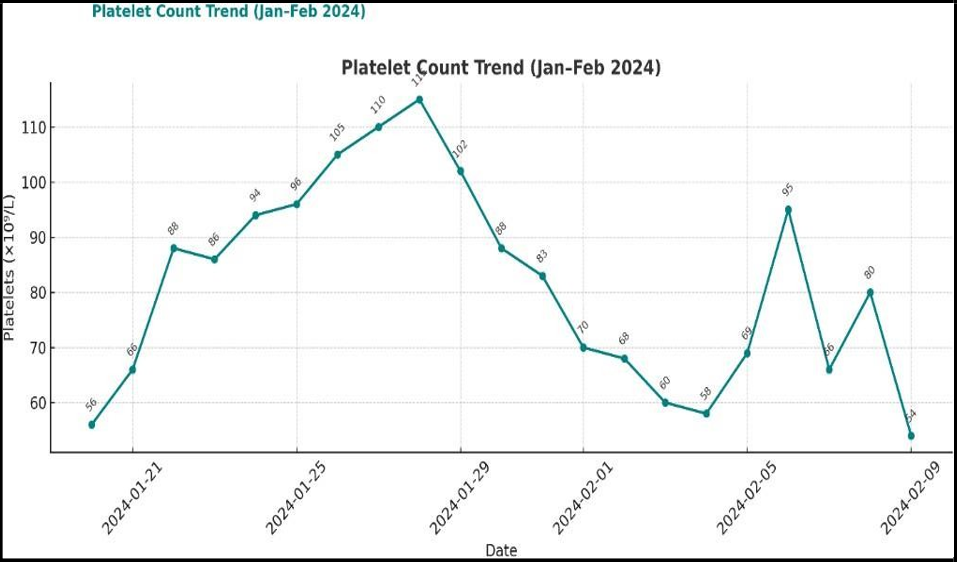 Figure 4: Platelet Count Trend from January till February 2024
Figure 4: Platelet Count Trend from January till February 2024
On March 26, 2024, the patient was readmitted due to acute graft dysfunction, evidenced by a rise in serum creatinine to 216 µmol/L following removal of the double-J (DJ) stent. A Doppler ultrasound of the renal transplant revealed new-onset mild hydronephrosis, raising concern for possible post-obstructive changes or ureteral compromise.
Despite this, graft perfusion remained satisfactory, with a patent main renal artery and vein. Peak systolic velocities were measured as follows:
1. Pre-anastomosis: 140 cm/s
2. Post-anastomosis: 131 cm/s
3. At the anastomosis site: 366 cm/s
These findings suggest post-anastomotic turbulence, possibly reflecting vascular remodeling or narrowing, though without evidence of critical stenosis. The interlobar and arcuate arteries demonstrated normal waveforms, with resistive index (RI) values ranging from 0.68 to 0.82, slightly elevated compared to prior values (0.62–0.74), but still within acceptable limits for post-transplant surveillance.
Importantly, there was significant regression of the perigraft fluid collection, now measuring 29 mL, indicating resolution of the previously noted postoperative accumulation.
A Tc-99m MAG3 renogram was performed to further assess graft function. The scan was abnormal, demonstrating slightly reduced perfusion and progressive parenchymal retention, consistent with graft dysfunction. Differential considerations included acute rejection, vasomotor instability, and drug-induced nephrotoxicity, warranting tissue sampling and close follow-up. The observed hydronephrosis was dilated but nonobstructive, likely attributable to recent stent removal and transient urodynamic changes [Figure 5].
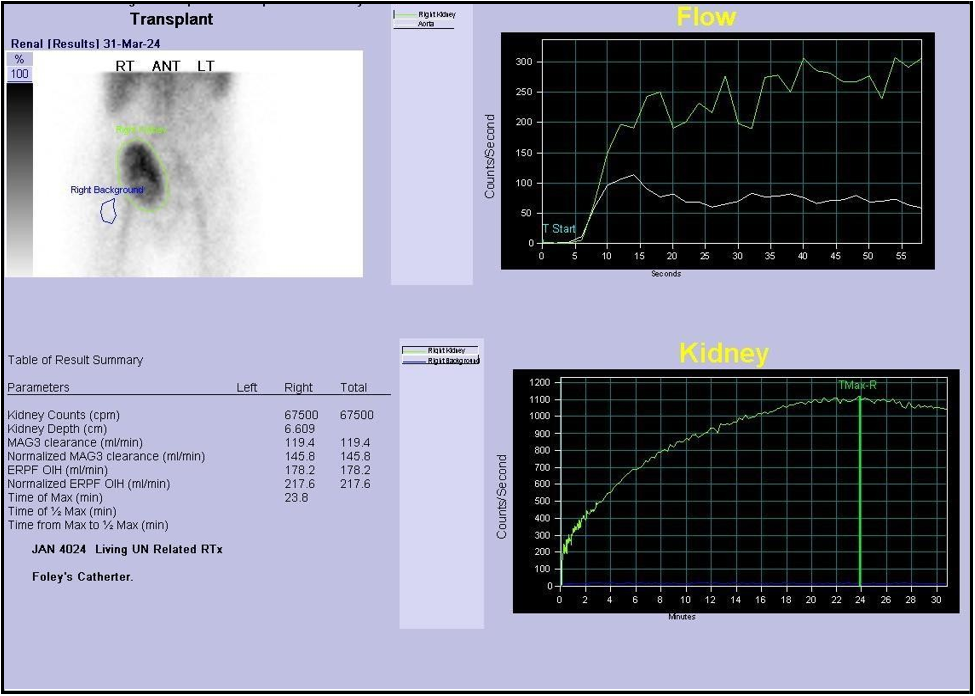 Figure 5: Tc-GGm renogram showing reduced perfusion with progressive parenchymal retention and dilated, non-obstructive hydronephrosis.
Figure 5: Tc-GGm renogram showing reduced perfusion with progressive parenchymal retention and dilated, non-obstructive hydronephrosis.
A repeat percutaneous kidney biopsy was performed and histopathologic analysis revealed 18 of 34 glomeruli globally sclerosed, indicating significant chronic glomerular injury. There were features consistent with urinary tract infection, and moderate peritubular capillaritis was noted, although definitive glomerulitis was absent, C4d stain negative.
A multidisciplinary team, including transplant nephrology, transplant surgery, urology, infectious diseases, and hematology, coordinated the patient’s care. Initial management involved placement of a foley catheter and the administration of meropenem, which was continued to ensure appropriate antimicrobial coverage until urine culture results were finalized. Blood and urine cultures grew pan-sensitive Klebsiella pneumoniae, confirming the infectious organism and informing ongoing therapy.
Following removal of the foley catheter, the patient’s serum creatinine rose markedly to 504 µmol/L. In response to this acute deterioration in renal function, a nephrostomy was placed on April 8, 2024. Imaging via nephrostogram showed a proximal ureteric contrast leak and absence of bladder opacification. This intervention was followed by an improvement in creatinine to 116.
On May 21, 2024, the patient was readmitted following two days of anuria due to a blocked nephrostomy tube. Ultrasound evaluation revealed that vascularity within the renal graft remained patent. However, there was a notable progression of the perigraft fluid collection, which had increased substantially to 849 cc from a previous measurement of 30 cc. Additional findings included moderate abdominal ascites and moderate hydronephrosis.
To address the obstruction, a nephrostomy tube exchange was performed, and fluid was drained from the perigraft collection (infected urinoma) [Figure 6]. Drainage cultures subsequently tested positive for ESBL-producing Escherichia coli. The patient was treated with meropenem in response to this infection. Following successful resolution of the infection, the patient underwent cystoscopy that showed complete occlusion of transplanted ureter, then she underwent surgery involving ureter-to-ureter anastomosis. In the postoperative period, the patient experienced additional episodes of urinary tract infection. The causative organisms included Pseudomonas aeruginosa, Klebsiella pneumoniae, and Enterobacter cloacae. All pathogens identified were sensitive to cefepime, allowing for targeted antimicrobial therapy and effective management of these recurrent infections. Subsequently, both the DJ stent and nephrostomy tube were removed.
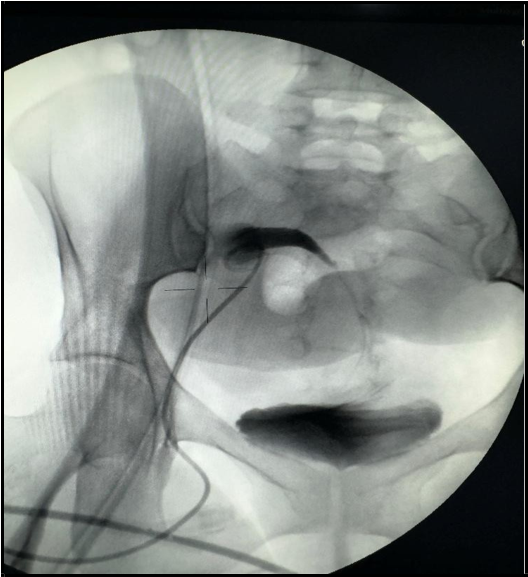
(A)
(B)
Figure 6: A) nephrostomy tube exchange, B) ultrasound guidance collection drainage
The patient’s most recent clinic visit was in October 2024, with laboratory values showing creatinine at 106 µmol/L, hemoglobin at 10.2 g/dL, and platelet count of 50×10⁹/L. After this visit, she relocated to the south of Saudi Arabia.
Discussion
Renal transplantation in MYH9-RD is feasible, but patients often face complex peri- and post-transplant challenges. In our patient, macrothrombocytopenia posed theoretical bleeding risks, yet she tolerated cesarean deliveries, multiple transfusions, kidney transplant operation, thymoglobulin, and invasive procedures without catastrophic hemorrhage. This observation is consistent with previous reports indicating that bleeding in MHA is often less severe than platelet counts suggest, as automated analyzers may underestimate platelet numbers, leading to exaggerated concerns about bleeding risk [11]. Nevertheless, repeated transfusions were required during her course, emphasizing the resource-intensive nature of perioperative support.
In addition to hematological challenges, this case highlights a significant immunologic risk. The patient was highly sensitized, likely from prior pregnancies and transfusions, and developed Banff 2A acute cellular rejection. She responded well to corticosteroids and thymoglobulin, reinforcing the importance of vigilant immunological monitoring and prompt therapy in sensitized MYH9-RD transplant recipients. The literature supports that rejection is not inherently more common in MYH9-RD, but sensitization can complicate donor matching and post-transplant outcomes [5, 6, 12].
Another notable feature of this case was the coexistence of a perigraft hematoma and Doppler findings suspicious for renal artery stenosis (RAS). Hematomas are relatively common in thrombocytopenic patients and can exert compressive effects that mimic or worsen vascular compromise. Differentiating between true RAS and compression by hematoma is challenging, particularly when invasive angiography is contraindicated due to bleeding risk. In our case, conservative monitoring with imaging was favored, and subsequent biopsy revealed acute rejection, underscoring the need to consider multiple concurrent mechanisms of graft dysfunction. Similar diagnostic dilemmas have been described in other MYH9-RD transplant recipients, where rejection, vascular compromise, and hemorrhagic complications may overlap and confound management [8, 9].
Recurrent urinary tract infections (UTIs) with multidrug-resistant organisms further complicated the post-transplant course. Our patient experienced infections with Klebsiella pneumoniae, ESBL Escherichia coli, Pseudomonas aeruginosa, and Enterobacter cloacae. Such infections are common in transplant tourism recipients, reflecting both the high burden of urological complications and exposure to resistant organisms [6]. They often require prolonged broad-spectrum antibiotics and multiple surgical interventions, as in our case with nephrostomy, ureteric reconstruction, and repeated stent exchanges. Despite these setbacks, long-term graft function was preserved, consistent with reports that nephropathy in MYH9-RD does not recur in the allograft, as the podocyte defect is intrinsic to native kidneys [9, 12].
The existing literature consists mainly of case reports and small series. Fabbian et al. described a patient with MHA in whom platelet counts were underestimated by automated analyzers [11]. Vischini et al. reported a transplant recipient with macrothrombocytopenia initially misdiagnosed as Epstein syndrome, who underwent two successful transplants [7]. Min et al. documented the first Korean case of transplantation in MYH9-RD with severe thrombocytopenia [8]. Zununi Vahed et al. reported a patient with MYH9-RD who developed a post-transplant hematoma but achieved stable long-term graft function [9]. Tabibzadeh et al. described a multicenter cohort of 13 genetically confirmed patients with heterogeneous renal outcomes, often misdiagnosed as immune thrombocytopenia or Alport syndrome; importantly, recurrence of nephropathy after transplant was not observed [12]. More recently, Nguyen et al. reported two patients with MYH9-associated nephropathy where severe thrombocytopenia precluded biopsy, highlighting the importance of genetic testing for diagnosis and prognosis [13]. These reports, together with ours, support that transplantation is generally successful in MYH9-RD, though perioperative bleeding and infectious complications remain major concerns.
Our case is unique in that it demonstrates the combined occurrence of perigraft hematoma, vascular turbulence, acute rejection, recurrent multidrug-resistant UTIs, and ureteric obstruction, all within the same patient. This underscores the need for individualized and multidisciplinary management, integrating nephrology, hematology, surgery, and infectious diseases. Long-term follow-up should also include monitoring for extrarenal manifestations such as hearing loss and cataracts, which may evolve independently of renal disease [2, 14].
Conclusion
Kidney transplantation is a viable treatment for end-stage kidney disease in MYH9-RD, but requires meticulous, multidisciplinary management. Our case demonstrates that even in the presence of macrothrombocytopenia, sensitization, vascular complications, and recurrent multidrug-resistant infections, favorable graft outcomes can be achieved. Clinicians should maintain a high index of suspicion for overlapping mechanisms of graft dysfunction—including rejection, vascular compromise, and hemorrhagic complications— and tailor diagnostic and therapeutic strategies accordingly.
Conflict of Interest
The authors have no conflicts of interest to disclose regarding this case report, its authorship, or its publication.
References
- Savoia A, Pecci A (2008) MYH9-Related Disease. In: Adam MP, Ardinger HH, Pagon RA, et al., editors. GeneReviews®. Seattle: University of Washington.
- Pecci A, Verver EJ, Schlegel N, Canzi P, Boccio CM, et al. (2014) Cochlear implantation is safe and effective in patients with MYH9- related disease. Orphanet J Rare Dis. 9: 100.
- Singh N, Nainani N, Arora P, Venuto RC (2009) CKD in MYH9- related disorders. Am J Kidney Dis. 54(4): 732–40.
- Drenckhahn D, Franke RP (1988) Ultrastructural organization of contractile and cytoskeletal proteins in glomerular podocytes of chicken, rat, and man. Lab Invest. 59(6): 673–82.
- Pecci A, Panza E, Pujol-Moix N, Klersy C, Di Bari F, et al. (2008) Position of nonmuscle myosin heavy chain IIA (NMMHC-IIA) mutations predicts the natural history of MYH9- related disease. Hum Mutat. 29(3): 409–17.
- Hama EY, Yamaguchi S, Uchiyama K, Kojima D, Nagasaka T, et al. (2023) Successful renal transplantation following hemodialysis as bridging therapy in a patient with Fechtner syndrome: a case report and literature review. Ren Replace Ther. 9: 50.
- Vischini G, Farneti F, Niscola P, Stefoni A (2012) Pregnancy desire unmasks a MYH9 syndrome in a kidney transplant woman. Saudi J Kidney Dis Transpl. 23(4): 834– 5.
- Min SY, Ahn HJ, Park WS, Kim JW (2014) Successful renal transplantation in MYH9-related disorder with severe macrothrombocytopenia: first report in Korea. Transplant Proc. 46(2): 654–6.
- Vahed SZ, Niknafs B, Noshad H, Tolouian R, Shoja MM, et al. (2023) Renal transplantation in a patient with MYH9-related disease; a case report. J Nephropathol. 12(1): e15980
- Sun J, Zhou L, He Q (2014) MYH9-related disease with nephropathy: clinicopathologic features in 11 Chinese patients. Kidney Blood Press Res. 39(6): 622–32.
- Fabbian F, Ricci F, De Giorgi A, Forcellini S, Scanelli G (2010) The May–Hegglin anomaly in a kidney transplant recipient. NDT Plus. 3(3): 312.
- Tabibzadeh N, Fleury D, Labatut D, Bridoux F, Lionet A, et al. (2019) MYH9- related disorders display heterogeneous kidney involvement and outcome. Clin Kidney J. 12(4): 494–502.
- Nguyen MD, Dileep G, Quizon M, Nguyen V, Demerci A, et al. (2024) May-Hegglin anomaly associated nephropathy: case series. SAGE Open Med Case Rep. 12: 1–5.
- Shen K, Chen T, Xiao M (2024) MYH9-related inherited thrombocytopenia: the genetic spectrum, underlying mechanisms, clinical phenotypes, diagnosis, and management approaches. Res Pract Thromb Haemost. 8: e102552.