


Charlie Robin, Christian Ankney, DO, Kylie Johnson, Gabi Swidler, Brandon Gordon, Jordan Schaefer, Santiago Candocia, MD, Robert Marsh, MD, Mark Talamonti, MD, Rupa Patil, MD, Jason Robin, MD*
North Shore University Health System, 2150 Pfingsten Ave Suite 1200, Glenview, IL 60025
*Corresponding Author: Jason Robin, MD, North Shore University Health System, 2150 Pfingsten Ave Suite 1200, Glenview, IL 60025.
Abstract
Background: Pancreatic neuroendocrine tumors are a diverse group of neoplasms which coexist with gastrointestinal carcinoids as the main histiotypes of gastrointestinal neuroendocrine tumors. Pancreatic neuroendocrine tumors are found primarily in the pancreas and upper small intestine. Although pancreatic neuroendocrine tumors share many features with gastrointestinal carcinoids, they are generally considered separately because of their different clinical syndromes (carcinoid syndrome vs. pancreatic neuroendocrine tumor syndromes) molecular pathogenesis, biological behaviors, and difference in response to antineoplastic treatments. Common pancreatic neuroendocrine tumors include gastrinomas, insulinomas, glucagonomas, VIPomas, GRFomas, ACTHomas, somatostatinomas, pancreatic neuroendocrine tumors causing carcinoid syndrome and those causing hypercalcemia (PTHrPomas). A pancreatic neuroendocrine tumor secreting renin resulting in hypertension is exceptionally rare and has not been previously reported.
Case Presentation: We present a 43-year-old female with a grade 2 neuroendocrine tumor diagnosed four years earlier, initially treated with distal pancreatectomy and splenectomy, who presented to her internist for routine follow up with a blood pressure of 150/100 mmHg. An MRI three months earlier showed no recurrent disease with stable Chromogranin A levels. A workup for secondary hypertension was pursued which discovered an elevation in renin and aldosterone levels without evidence of hypercoagulability or compromised vascular supply to the kidneys. Four months later, an MRI showed a new metastatic lesion in the liver and the Chromogranin A levels increased from her baseline. With two microwave ablations and Lanreotide, her blood pressure normalized as did her renin and aldosterone levels.
Conclusion: Newly diagnosed hypertension in a patient with a history of a pancreatic neuroendocrine tumor should prompt a workup for secondary hypertension as this may be a sign of tumor recurrence. Hyperreninemia in the setting of a neoplasm has mostly been described with juxtaglomerular cell tumors of the kidney and has never been described with a pancreatic neuroendocrine tumor. Treating the underlying tumor can eliminate the overproduction of renin and normalize the blood pressure.
Keywords: Pancreatic neuroendocrine tumors, hypertension, hyperreninemia, secondary hyperaldosteronism, renin secreting tumor
Introduction
Background
Pancreatic neuroendocrine tumors (PNETs) comprise gastrointestinal carcinoids as the main groups of gastrointestinal neuroendocrine tumors. PNETs are neuroendocrine tumors that are found primarily in the pancreas and upper small intestine [1]. Although pancreatic neuroendocrine tumors share features with gastrointestinal carcinoids, they are considered separately because of their different clinical syndromes (carcinoid syndrome vs. PNET syndromes) and their different molecular pathogenesis, biological behaviors, and response to anti-neoplastic treatments [2,3-6].
There are ten different recognized PNETs. Nine of these are associated with a specific clinical syndrome including gastrinomas (Zollinger-Ellison syndrome), insulinomas, glucagonomas, VIPomas (Vasoactive Intestinal Peptide or Verner-Morrison syndrome, pancreatic cholera), GRFomas (Growth Hormone releasing Factor), ACTHomas (Adrenocorticotropic), somatostatinomas, PNETs causing carcinoid syndrome and Parathyroid Releasing Hormone Peptide or PTHrPomas. One PNET syndrome is not associated with a specific hormonal syndrome and is frequently referred to as a nonfunctional PNET (NF-PNET) [2,7].
In addition to these PNET subtypes, there are other very rare PNETs with clinical syndromes in which only a few cases have been reported. These include PNETs ectopically secreting erythropoietin resulting in erythroblastosis; PNETs secreting GLP-1 (Glucagon Like Peptide) or IGF-2 (Insulin Growth factor) which is associated with hypoglycemia; and PNETs secreting lutenizing hormone causing masculinization [8-11]. Ruddy et al described hypertension associated with a renin-secreting tumor of the pancreas, but the underlying pathology was adenocarcinoma [12]. To our knowledge, this is the first case report of a PNET presenting with hypertension, likely due to hyperreninemia.
Case presentation
In the September of 2020, a 43-year-old female with a medical history of a PNET presented to her internist for routine follow up and was noted to be significantly hypertensive with a blood pressure of 150/100 mmHg. She was diagnosed with the PNET in 2016 and she was resected at that time. Her pathology conveyed a grade 2 tumor that was 8.5 cm in size. There was focal splenic and vascular invasion. Six peri-pancreatic lymph nodes were negative for metastasis (0/6). The margins in the pancreas were free of tumor. Due to the splenic invasion, she also underwent splenectomy at that time. Her medical history was also notable for four miscarriages.
Regarding her PNET, she had been followed with serial chromogranin A levels since her surgery which had been in the normal range (range 46-59 ng/ml, September 2020 level 48 ng/ml, normal < 93 ng/ml, see table 1). In addition, she had serial imaging with MRIs of her abdomen with contrast every six months since the surgery. Her most recent MRI was in June 2020, and it conveyed stable postoperative changes from the prior distal pancreatectomy and splenectomy with no evidence of new or recurrence of disease.
At the time of her September 2020 office visit, aside from the hypertension, her vitals and exam were unremarkable. She was noted to be 5' 5" (1.651 m) weighing 68.9 kg (152 lb) with a BMI of 25.29 kg/m², a heart rate of 70 beats per minute with a normal cardiac and abdominal exam. Her internist had her monitor her blood pressures at home which ranged 150-160 /90-100 mmHg. She reported some intermittent headaches at home. Based on the unusual presentation of hypertension in an otherwise healthy 43-year-old female, a workup for secondary hypertension was undertaken. Her basic metabolic panel conveyed:
Na 137 meq/L
K 4.3 meq/L
Cl 99 meq/L
CO2 27 meq/L
BUN 9 mg/dL
Creatinine 0.8 mg/dL
Glucose 87 mg/dL
24-hour urine total metanephrines 282 mcg (normal 119-451) 24-hour urine 5H1AA 3.9 mg (normal <7.3)
Plasma Aldosterone 60 ng/dl (normal <21)
Plasma Renin 12 ng/ml (normal 0.6 to 3.0)
A renal artery ultrasound showed no evidence of stenosis, and a CT of the aorta showed no evidence of coarctation. In the setting of multiple miscarriages and new onset hypertension, a workup for Anti Phospholipid Syndrome was performed which was unremarkable (Lupus Anticoagulant, Beta 2 Glycoprotein and Anticardiolipin Antibodies were all within normal limits).
A low dose of lisinopril (2.5 mg) was initiated which had no impact on her blood pressure but was associated with extreme fatigue and lightheadedness so it was stopped within a week. She was subsequently started on losartan 50 mg which lowered her blood pressure to the low 140s/high 80s. A repeat Aldosterone and Renin levels was checked while on losartan:
Plasma Aldosterone 34 ng/dl (normal < 21) Plasma Renin 79 ng/ml (normal 0.6 to 3.0)
Soon after starting the losartan, she again experienced fatigue, so this too was stopped. Several weeks later while off of blood pressure therapy, repeat Aldosterone and Renin levels were again obtained.
Plasma Aldosterone 32 ng/dl (normal < 21) Plasma Renin 9.3 ng/ml (normal 0.6 to 3.0)
The patient continued to feel well off medication for blood pressure yet remained hypertensive. She returned for a routine surveillance MRI in February of 2021 which revealed a T2 hyperintense, T1 hypointense lesion with restricted diffusion measuring 9 x 11 mm in the posterior right hepatic lobe (see image 1). This finding was concerning for new metastatic disease. Her Chromogranin A level also rose to 83 ng/ml (see table 1).
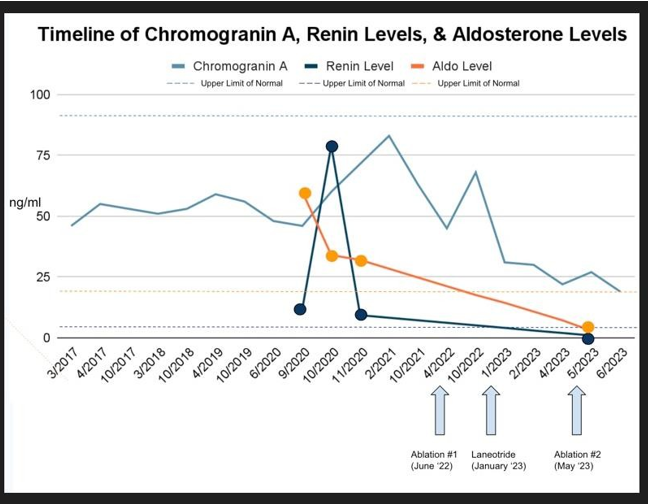
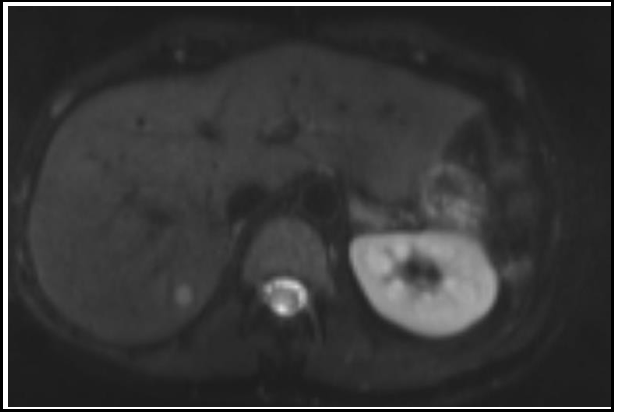
Figure 1: MRI Abdomen February 2021
New T2 hyperintense, T1 hypointense lesion with restricted diffusion measuring 9 x 11 mm in the posterior right lobe of the liver.
The lesion was felt to be small and unlikely to be seen on PET at that time. She was sent for a needle biopsy which was non-diagnostic with unremarkable hepatic parenchyma. At this time, her blood pressure remained elevated at 150/100 mmHg, and she started on amlodipine 5mg plus losartan 50 mg which she was able to tolerate. This lowered her blood pressure to the low 130s/80s.
She had a follow up MRI in April 2022, and the lesion grew to 18 x 11 mm (Image 2).

Figure 2: MRI Abdomen April 2022
Increasing size of T2 hyperintense, T1 hypointense lesion in the posterior right hepatic lobe (11 x 18 mm)
She subsequently completed a gallium DOTATATE PET scan on May 5th, 2022. This showed that the hepatic lesion within the right posterior segment of the liver was DOTATATE acid and was suggestive of a metastatic neuroendocrine cancer lesion. No other evidence of abnormal DOTATATE uptake was seen (Image 3). Interestingly, her Chromogranin A levels came down to her baseline of 45 ng/ml without intervention (see table 1).
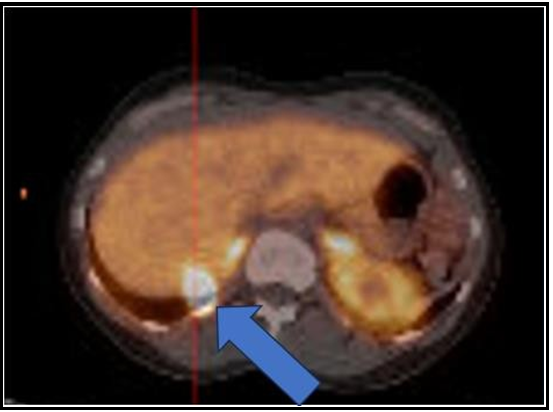
Figure 3: PET/CT 68 gallium DOTATATE study May 2022
The hepatic lesion within posterior right hepatic lobe is DOTATATE avid and is suggestive for a metastatic neuroendocrine lesion
She was seen by both her surgeon and an interventional radiologist who felt she was an excellent candidate for percutaneous thermal ablation of the mass. On June 18th, 2022, she underwent ultrasound and CT guided microwave ablation of the mass with a 15 cm thermosphere antenna for 4 minutes at 75 watts for an ablation zone of 2.9 cm x 3.2 cm. MRI of the abdomen three weeks later on July 10th, 2022, demonstrated successful ablation of the right liver lobe mass, without evidence of residual or recurrent tumor.
Follow up Chomogranin A levels in October 2022 had risen again to 68 ng/dL (see table 1). A follow up MRI in October 2022 showed what was thought to be successful ablation therapy of the lesion in the posterior segment of the right lobe of the liver with interval decreased size of the cavity. However, there was a new sub-centimeter enhancing lesion in the medial segment of the left lobe for new metastasis (Image 4). Her blood pressure remained in the in the 130s/80s while on losartan and amlodipine.
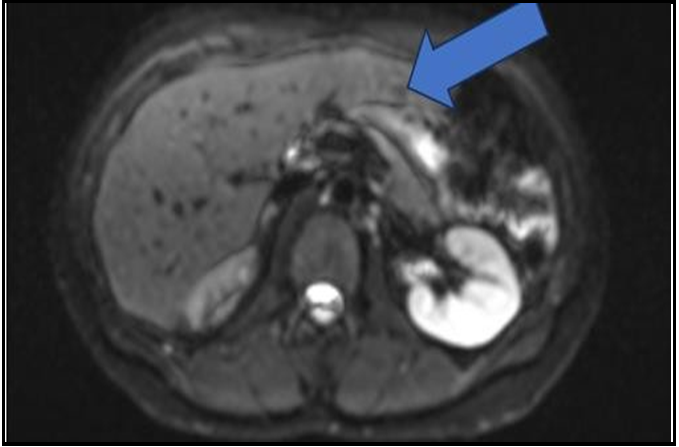
Figure 4: MRI Abdomen October 2022
Successfully ablation therapy of the lesion in the posterior segment of the right lobe of the liver with interval decreased size of the cavity. However, there is a new sub centimeter enhancing lesion in the medial segment of the left lobe consistent with a new metastasis.
A repeat DOTATATE PET scan on 10/21/2022 conveyed persistent intense activity in the distribution of the right posterior hepatic lobe lesion worrisome for residual/recurrent neoplasm. There was no significant activity seen in the distribution of the new arterial enhancing lesion of recent MRI from October 2022. However, it was felt that the lesions was too small for PET/CT characterization (image 5)
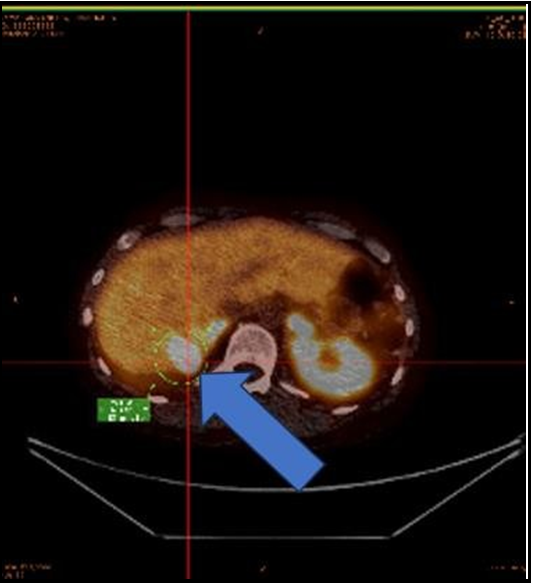
Figure 5: PET/CT 68gallium dotatate study October 2022
Intense activity in the distribution of the right posterior hepatic lobe lesion consistent with residual/recurrent neoplasm
After the PET which demonstrated intense activity in the distribution of the right posterior hepatic lobe, she had follow-up with Interventional Radiology who felt that DOTATATE scan uptake in the right lobe was out of proportion to the lesion size and that the new hyper-enhancing metastatic lesion in the left lobe was too small to be seen on DOTATATE. After discussion with the oncologist, it was felt that Somatostatin therapy was the best course in case she was progressing or not responding to localized therapy.
In January 2023 she was started on Lanreotide injections every 28 days. Immediately after her first injection, her Chromogranin A levels fell to the 30s and her blood pressure was noted to be in the 100s/50s (see table 1). It was at this time she discontinued blood pressure therapy (losartan 50 mg and amlodipine 5 mg) as she was lightheaded. A follow up MRI in April 2023 showed post procedural change following ablation with an ablation cavity measuring 1.2 x 1.0 cm (previously 1.2 x 1.1 cm). However, there was a small amount of internal nodular enhancement (see figure 6). The previously reported lesion in the medial segment left lobe on October 16th, 2022, was not seen and thought to be related to a perfusion anomaly.
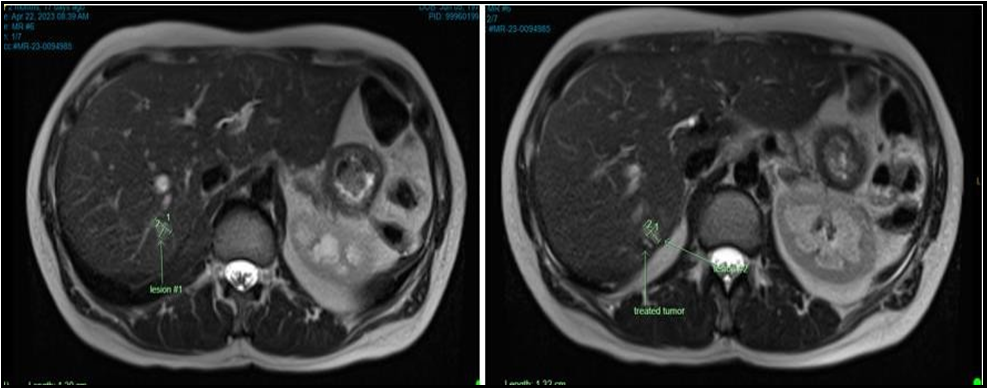
Figure 6: MRI Abdomen April 2023
Lesion #1: Immediately adjacent to the ablation cavity is an area of diffusion restriction with abnormal T1 hypo-/T2 isointense signal with associated arterial and perilesional enhancement; the cranial component measures 1.3 x 0.9 cm.
Lesion #2: The caudal medial component measures 1.3 x 0.8 cm.
Previously reported lesion in the medial segment left lobe on 10/16/2022 is again not seen and perhaps related to a perfusion anomaly.
The decision was made to re-ablate the lesions of the right lobe which were completed on May 13, 2023, where she had ultrasound and CT-guided microwave ablation of two right hepatic lobe metastases. After the ablation she remained normotensive without the need for blood pressure therapy. Her Chromogranin A levels continued to fall. A repeat Renin and Aldosterone assessment was done showing (see table 1):
Plasma Aldosterone 2.9 ng/dl (normal <21)
Plasma Renin 0.96 ng/ml (normal 0.6 to 3.0)
At the time of this report, she is three months out from her second ablation, and she remains on Lanreotide injections. Her blood pressure remains well controlled in the 100/50 mmHg range without blood pressure medication and her Chromogranin A levels continue to decline.
Discussion
The key management steps for all PNETs include suspect the diagnosis, establish the diagnosis, determined whether an inherited disorder is also present, control the hormone-excess state, establish the primary tumor location with extent of disease, surgical resection of the tumor, if possible, systemic treatment if malignant and unresectable, and finally the long-term follow-up of the patient [2]. All functional PNETs have two treatment considerations: treatment is required of the hormone-excess state and treatment needs to be directed at the PNET itself, because in all cases except for insulinomas (< 10 % malignant) the PNET show malignant behavior [13,14]. In older studies where effective treatments did not exist, many patients with functional PNETs died of the uncontrolled hormone-excess state, demonstrating the importance of control of this state both acutely and long-term. Increasingly, at present PNET patients are dying of the malignant behavior of these tumors, demonstrating the importance of surgical removal of the PNET, whenever possible and effective control of the tumor in patients with advanced malignant disease [13,14]. Unfortunately, in many cases the diagnosis is delayed, and the patient presents with advanced metastatic disease making surgical cure impossible.
In this case, the first manifestation of tumor recurrence was the unexplained acute onset of stage II hypertension in an otherwise healthy woman with a BMI of 25.29 kg/m². Based on her stable Chromogranin A levels and recent benign MRI, tumor recurrence was not highly considered. It was appropriate to pursue a secondary workup for hypertension based on its sudden onset.
In this case, the combination of elevated aldosterone and elevated renin levels brought forth a diagnosis of secondary hyperaldosteronism. The differential diagnosis with secondary hyperaldosteronism is narrow and includes coarctation of the aorta, renal artery stenosis, arterial hypercoagulable states with micro- thrombi formation in the afferent arterioles of the glomerulus and renin secreting tumors. The former diagnoses were all ruled out with imaging and assessment of Lupus Anticoagulant, Beta 2 Glycoprotein and Anti-Cardiolipin Antibodies which were all negative. Hypercoagulable states causing primarily venous thrombosis were not considered relative in this clinical context [17].
Most often, tumors associated with secondary hyperaldosteronism are benign juxtaglomerular renin secreting tumors. These tumors must be differentiated from hypereninemia from other tumors such as nephroblastoma and renal cell carcinoma which represent the next most common etiologies. Strick diagnostic criteria that the tumor cells are involved in ectopic renin secretion include measurement of plasma and tumoral tissue renin activity, absence of any other cause of hyperreninemia, regression of hyperreninemia with tumor resection, and demonstration of renin antigen in the tumor by immunohistochemistry or in situ hybridization with human renin probes [18].
Unfortunately, in our case, upon tumor recurrence, tissue could not be obtained, yet the diagnosis of secondary hyperaldosteronism due to the PNET is supported by the significant drop in blood pressure after the initiation of Lanreotide in conjunction with the second ablation procedure. Chronologically, it was the systemic treatment which had the greatest impact, likely by suppressing renin secretion. This occurred in parallel with a steep drop in Chromogranin A levels (see figure 1). When she had clinical evidence of metastatic disease (elevated Chromogranin A levels and positive PET imaging) she required two blood pressure medications to keep her blood pressure in the 130’s/80s range. After treatment with Lanreotide, she remained normotensive in the 100s/50s without blood pressure therapy. Finally, a repeat renin and aldosterone assessment while off of blood pressure therapy conveyed nearly undetectable levels compared to her levels in September and October of 2020 (see figure 1).
Hyperaldosteronism was first described in 1954 by Dr. Jerome Conn, who noted a syndrome of periodic muscle weakness, hypokalemia, alkalosis, and hypertension with elevated urinary aldosterone levels [15]. Subsequently, hypokalemia came to be viewed as an important component of this condition, to the point that it became almost a sine qua non for the diagnosis. Although hyperaldosteronism was classically suspected in patients presenting with the triad of hypertension, hypokalemia, and metabolic alkalosis, this phenotype likely describes an extreme form of the condition. In fact, only 9 % to 37 % of patients with hyperaldosteronism have hypokalemia, and the most common presentation is normokalemic hypertension [16]. Hypokalemia often alerts the clinician to a possible diagnosis of aldosteronism; however, the absence of hypokalemia holds a poor negative predictive value for diagnosis and should not be used to exclude the presence of the disorder. In the case of secondary hyperaldosteronism, hypertension is not only attributed to by the elevated aldosterone state, but also by the increased levels of angiotensin II which leads to vasoconstriction.
In regard to her renin and aldosterone levels, it is important to recognize that she was considered well hydrated and volume replete prior to each laboratory assessment. This was assessed clinically and can also be demonstrated in her BUN: Creatinine ratio which would have been elevated had the elevated renin level been secondary to hypovolemia. Her initial levels were significantly elevated and were drawn prior to the initiation of blood pressure therapy (see table 1). Thus, the elevation cannot be blamed on pharmacologic interference of the renin-angiotensin-aldosterone system (RAAS) pathway. Her second assessment was done while she was on the angiotensin receptor block (ARB), losartan. As expected, the renin levels increased significantly although more so than usually seen in the setting of downstream suppression of Angiotensin II levels. In addition, the aldosterone levels remained elevated which would not be expected while on an ARB. Her third evaluation while off of pharmacotherapy again showed elevation in both the renin and aldosterone levels consistent with secondary hyperaldosteronism. The final values after the second ablation and initiation of Lanreotide conveyed normalization of both renin and ldosterone which also corresponded to both a significant drop in blood pressure and her lowest Chromogranin A levels. This strongly supports that the treatment of the metastatic lesion(s) to the liver was paramount in controlling the blood pressure and suppressing renin secretion.
A final consideration in this case is that she did not present with hypertension upon her initial presentation in 2016. It has been well described that PNETs may evolve and cause hormonal hypersecretion–related symptoms that were not present at the initial diagnosis, termed metachronous hormonal syndromes (MHSs). In one case series, MHSs developed after a median of 55 months which is a similar time frame as described in our case report [19]. Changing biologic behavior of PNETS may explain this phenomenon [20].
Conclusions
In conclusion, we describe the first case of a 43-year-old woman with a history of a PNET whose presentation of hypertension was her first manifestation of recurrence. Based on her pre-treatment renin and aldosterone levels and the reduction in levels and blood pressure after initiation of Lanreotide as well as the second ablation, her tumor was likely a renin secreting tumor. Patients with PNETs with new onset hypertension should be evaluated for secondary hyperaldosteronism. The renin and aldosterone levels in this case will be used for future monitoring.
List of abbreviations
PNET: Pancreatic neuroendocrine tumors VIP: Vasoactive Intestinal Peptide
GRF: Growth hormone releasing factor secreting. ACTH: Adrenocorticotropic
PTHrP: Parathyroid Releasing Hormone Peptide NF: nonfunctional
GLP-1: Glucagon like peptide IGF-2 Insulin Growth factor BPM: Beats per minute
BMI: Body mass index
RAAS: renin-angiotensin-aldosterone system ACE angiotensin converting enzyme.
ARB angiotensin receptor blocker
Declarations
Ethics approval and consent to participate.
The ethics committee of North Shore University Health System approves the submission of this manuscript.
Consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images. A copy of written consent is available for review by the Editor in Chief of this journal (see below).
Availability of data and materials: Not applicable
Competing interests: We, the authors of this manuscript, declare that we have no competing interests.
Funding: None
Acknowledgements: None
Authors' contributions
CR analyzed and interpreted the patient data regarding trend in Chromogranin A levels, created table 1 and was a major contributor in writing the manuscript.
CA researched pertinent literature, interpreted the pathology, and was a major contributor in writing the manuscript.
GS researched the pertinent literature, interpreted the hormonal levels and was a major contributor in writing the manuscript.
BG researched pertinent literature, interpreted the hematologic data, and was a major contributor in writing the manuscript.
JS researched the pertinent literature, interpreted the hematologic data, and was a major contributor in writing the manuscript.
SC was the primary care physician who initiated the work up and was a major contributor in writing the manuscript.
RM was the treating oncologist and was a major contributor in writing the manuscript.
MT was the treating surgical oncologist and was a major contributor in writing the manuscript.
RP was a consulting cardiologist and was a major contributor in writing the manuscript.
JR was the treating cardio oncologist, reviewed the paper, and was a major contributor in writing the manuscript.
References
- Oberg K (2010) Pancreatic endocrine tumors. Semin Oncol. 37(6): 594–618.
- Metz DC, Jensen RT (2008) Gastrointestinal neuroendocrine tumors: Pancreatic endocrine tumors. Gastroenterology. 135(5): 1469–1492.
- Kloppel G (2011) Classification and pathology of gastroenteropancreatic neuroendocrine neoplasms. Endocr Relat Cancer. 18 (Suppl 1): S1–S16
- Pavel M, Baudin E, Couvelard A, Krenning E, Oberg K, et al. (2012) ENETS Consensus Guidelines for the Management of Patients with Liver and Other Distant Metastases from Neuroendocrine Neoplasms of Foregut, Midgut, Hindgut, and Unknown Primary. Neuroendocrinology. 95(2): 157–176.
- Kulke MH, Benson AB 3rd, Bergsland E, Berlin JD, Blaszkowsky LS, et al. (2012) Neuroendocrine tumors. J Natl Compr Canc Netw. 10(6): 724–764.
- Chen M, Van Ness M, Guo Y, Gregg J (2012) Molecular pathology of pancreatic neuroendocrine tumors. J Gastrointest Oncol. 3(3): 182–188.
- Jensen RT, Cadiot G, Brandi ML, de Herder WW, Kaltsas G, et al. (2012) ENETS Consensus Guidelines for the Management of Patients with Digestive Neuroendocrine Neoplasms: Functional Pancreatic Endocrine Tumor Syndromes. Neuroendocrinology. 95(2): 98–119.
- Samyn I, Fontaine C, Van Tussenbroek F, Pipeleers-Marichal M, De Greve J (2004) Paraneoplastic syndromes in cancer: Case 1. Polycythemia as a result of ectopic erythropoietin production in metastatic pancreatic carcinoid tumor. J Clin Oncol. 22(11): 2240–2242.
- Roberts RE, Zhao M, Whitelaw BC, Ramage J, Diaz-Cano S, et al. (2012) GLP-1 and Glucagon Secretion from a Pancreatic Neuroendocrine Tumor Causing Diabetes and Hyperinsulinemic Hypoglycemia. J Clin Endocrinol Metab. 97(9): 3039–3045.
- Chung JO, Hong SI, Cho DH, Lee JH, Chung DJ, et al. (2008) Hypoglycemia Associated with the Production of Insulin-like Growth Factor II in a Pancreatic Islet Cell Tumor: A Case Report. Endocr J. 55(3): 607-12.
- Piaditis G, Angellou A, Kontogeorgos G, Mazarakis N, Kounadi T, et al. (2005) Ectopic bioactive luteinizing hormone secretion by a pancreatic endocrine tumor, manifested as luteinized granulosathecal cell tumor of the ovaries. J Clin Endocrinol Metab. 90(4): 2097–2103.
- Brignardello E, Manti R, Papotti M, Allìa E, Campra D, et al. (2004) Ectopic secretion of LH by an endocrine pancreatic tumor. J Endocrinol Invest. 27(4): 361–365.
- Ruddy MC, Atlas SA, Salerno FG (1982) Hypertension associated with a renin-secreting adenocarcinoma of the pancreas. N Engl J Med. 307(16): 993–997.
- Mignon M, Colombel JF (1999) Recent advances in pathophysiology and management of inflammatory bowel diseases and digestive endocrine tumors. Paris, France: John Libbey Eurotext Publishing Co. 192–219.
- Ito T, Igarashi H, Jensen RT (2012) Therapy of metastatic pancreatic neuroendocrine tumors (pNETs): recent insights and advances. J Gastroenterol. 47(9): 941–960.
- Conn JW (1955) Presidential address. I. Painting background. II. Primary aldosteronism, a new clinical syndrome. J Lab Clin Med. 45(1): 3–17.
- Funder JW, Carey RM, Mantero F, Murad MH, Reincke M, et al. (2016) The management of primary aldosteronism: case detection, diagnosis, and treatment: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 101(5): 1889–1916.
- Hamidou MA, Moreau A, Jego P, Testa A, Banisadr F, et al. (1995) Captopril and aspirin in treatment of renal microangiopathy in primary antiphospholipid syndrome. Am J Kidney Dis. 25(3): 486–488.
- Gaudemar M, Bruneval P, Camilleri JP (1988) Tumor syndromes with inappropriate renin secretion. Diagnostic criteria and review of published cases Ann Path. 8(2): 83-90.
- de Mestier L, Hentic O, Cros J, Walter T, Roquin G, et al. (2015) Metachronous hormonal syndromes in patients with pancreatic neuroendocrine tumors: a case series study. Annals of Internal Medicine. 162(10): 682-9.
- Alexandraki KI, Spyroglou A, Kykalos S, Daskalakis K, Kyriakopoulos G, et al. (2021) Changing biologic behavior of NETs during the evolution of the disease: progress on progression Enocrine Related Cancer. 28(5): 121-140.