


Hedström Yvette1,2, Larsson Lars1,2,3*
1Basic and Clinical Muscle Biology, Department of Physiology and Pharmacology, Karolinska Institute, Stockholm, Sweden.
2Center for Molecular Medicine, Karolinska Institute, Sweden
3Viron Molecular Medicine Institute, One Boston Place, 6th Floor, Boston, MA 02108, USA
*Corresponding Author: Larsson Lars, Basic and Clinical Muscle Biology, Department of Physiology and Pharmacology, Karolinska Institute, Stockholm, Sweden, Center for Molecular Medicine, Karolinska Institute, Sweden, Viron Molecular Medicine Institute, One Boston Place, 6th Floor, Boston, MA 02108, USA.
Abstract
Critical Illness Myopathy (CIM) is a serious and common complication of modern critical care, prolonging ICU and post-intensive care with negative consequences for patient quality of life and associated with increased morbidity and mortality. CIM has for many years been misdiagnosed as an acquired neuropathy due to misinterpretation of electrophysiological signals. CIM is a primary acquired myopathy characterized by a preferential loss of the molecular motor in skeletal muscle (myosin) resulting in severe muscle wasting and loss of muscle function in limb and trunk muscles. The preferential myosin loss is therefore used as a diagnostic criterion when separating myogenic from neurogenic pathophysiology of acquired muscle paralysis in long-term mechanically ventilated and immobilized ICU patients by quantifying myosin vs. actin content, i.e., the myosin actin ratio since thin filament proteins are less affected. However, it remains unknown if there is a redistribution of myosin and actin between the myofibrillar and cytoplasmic compartments, which will have an impact on muscle function and recovery. This study was therefore undertaken to quantify myosin and actin content in the myofibrillar and sarcoplasmic compartments using a unique experimental intensive care unit model perfectly mimicking the intensive care unit (ICU) condition with immobilization and mechanical ventilation during the development of CIM and the preferential myosin loss. Results from this study show no redistribution of myosin and actin between the different compartments during the progression of the preferential myosin loss, supporting the use of myosin vs. actin content as a diagnostic and prognostic marker of CIM.
Introduction
Critical Illness Myopathy (CIM) is a common consequence of modern critical care with dramatic negative consequences for patient quality of life, morbidity/mortality, and health care costs. CIM is characterized by paralysis and severe muscle wasting of limb and trunk muscles while craniofacial muscles are spared or less affected as well as sensory and cognitive functions [1-4]. If the patient survives critical illness, the prognosis is often good with re- expression of myosin and recovery of muscle mass and function [4- 6].
CIM has for many years been misdiagnosed as an acquired critical illness neuropathy (CIN) due to misinterpretation of electrophysiological signals showing near normal motor and sensory nerve conduction velocities, but very low compound muscle action potentials and interpreted as a selective motor neuron axonopathy, i.e., CIN [1-4].
However, low compound motor action potentials are frequently, but not always, observed also in CIM due to a decreased muscle membrane excitability as part of a primary myopathy [1-4,7]. CIM is more common than CIN but there are specific current treatment besides supportive interventions. Despite this, it is important to make as correct diagnosis as possible since specific treatments targeting underlying mechanisms have been identified in experimental studies and are currently being translated to clinical studies. The need for correct diagnosis is also emphasized by differences in prognosis between CIM and CIN, and pharmacological interventions frequently used in the intensive care such as steroids may have different effects between CIM and CIN. Finally, all patients deserve as correct a diagnosis as possible.
The hallmark of CIM is the preferential complete or partial loss of myosin and myosin associated proteins while thin filament proteins are spared or less affected, resulting in low myosin: actin ratios. Electrophoretic separation of myofibrillar proteins and the calculation of the myosin: actin ratio has therefore been used as a diagnostic CIM marker. In our group we originally used muscle samples obtained with percutaneous muscle biopsy instruments [4-6,8] and more recently using disposable microbiopsy instruments [7,9]. The smaller muscle quantity obtained with the microbiopsy (4-15mg) shows identical diagnostic precision as when analyzing the larger samples (80-100mg) obtained with the percutaneous conchotome method [7]. The preferential myosin loss is the result of transcriptional downregulation of myosin synthesis and activation of different proteolytic pathways, documented in both clinical and experimental studies in patients and animals exposed to more than five days of immobilization and mechanical ventilation [5,9-11]. However, contractile proteins can be expressed in the myofibrillar and sarcoplasmic pool and it remains unknown if there is a redistribution of myosin and actin between these pools during the progression of CIM.
Differences in the distribution of myosin and actin between the myofibrillar and sarcoplasmic pools may have an impact on the diagnostic precision of the myosin: actin ratio as well as being anticipated to have an impact on muscle function as well as the recovery from CIM. The present study was therefore undertaken to determine differences in the expression of myofibrillar vs. sarcoplasmic myosin and actin expression during the progression of CIM in an established experimental ICU model, i.e., a model allowing long-term studies of rats mechanically ventilated and immobilized (the longest duration a rat has been followed in this model to date is 96 days). We have previously shown that rats exposed to the ICU condition for durations longer than 5 days develop the CIM geno- and phenotype with preferential myosin loss coupled to transcriptional downregulation of protein synthesis and activation of proteolytic pathways according to a specific temporal pattern [11]. In the current study, we have reanalyzed the myosin and actin expression in the myofibrillar and sarcoplasmic pools in a distal hindlimb muscle from controls and rats exposed to immobilization for 5 days and longer using the optimized protocol to isolate sarcoplasmic and myofibrillar protein fractions presented by Roberts et al. [12].
Materials & Methods
Adult female Sprague Dawley rats were included in this study. The tibialis anterior (TA) muscle tissue was analyzed in 0-day controls (n=9), and rats exposed to immobilization and mechanical ventilation for 5 days (n=9), 8 days (n=8), 10 days (n=5), and 13-14 days (2-week group, n=6). There was no significant difference in initial body weights between controls (299±21g), 5-day (307±36 g), 8-day (304±46 g), 10-day (304±42 g), and 2-week (316±31 g) groups. The ethical committees at Uppsala University and Karolinska Institutet approved all aspects of this study.
Experimental ICU model: All animals were maintained in fluid and nutritional balance throughout the duration of the experimental procedures by introducing: 1) intra-arterial solution (0.6 ml/h) containing 21 ml H2O, 24 ml 0.5 N lactated Ringer, 0.84 g oxacillin Na, 0.65 mg α-cobrotoxin, 0.3 mg vitamin K (Synkavite), 20 meq K+ (as KCl); 2) an intra-venous solution (0.6 ml/h) containing 26 ml H2O, 16 ml 0.5 N lactated Ringer, 20% glucose (Baxter, Deerfield, IL, USA), 0.32 g oxacillin Na for the initial 24, then 8.5% Travasol amino acids (Baxter) and 20% Intralipid (Kabi, Uppsala, Sweden) were added subsequently to provide adequate nutrients [13,14]. Body temperature, peripheral perfusion and oxygen saturation (measured continuously with an infrared probe in a hind limb paw, MouseSTAT, Kent Scientific corp., Torrington, CT, USA) were monitored and maintained in the physiological range. The sham-operated controls were anesthetized with isoflurane, maintained in spontaneous breathing, received intravenous and intra-arterial solutions, and sacrificed within 2 hours of the initial isoflurane anaesthesia and surgery.
During surgery or any possible irritating manipulation, the anaesthetic isoflurane level, i.e., the Minimum Alveolar Concentration (MAC), was > 1.5%, which maintains the following states: 1) The electroencephalogram (EEG) was synchronized and dominated by high-voltage slow-wave activity; 2) mean arterial pressure, 90-100 mmHg, heart rate maintained below 420 beats/min; and 3) no evident EEG, blood pressure or heart rate responses to surgical manipulation. Isoflurane was delivered into the inspiratory gas stream by a precision mass-flow controller. After the initial surgery, isoflurane was gradually lowered (over 1-2 days) and maintained at MAC < 0.5% during the remaining experimental period. Rats were ventilated through a coaxial tracheal cannula at 72 breaths/min with an inspiratory and expiratory ratio of 1:2 and a minute volume of 180- 200 ml and gas concentrations of 40% O2, 56.5% N2 and 3% CO2, delivered by a precision (volume drift < 1%/ wk) volumetric respirator. Airway pressure was monitored continuously as well as end-tidal CO2 (EtCO2) and normocapnic condition maintained (EtCO2 = 37- 45 mmHg) as well as normoxia (SpO2 > 90%). Intermittent hyperinflations (6 per hour at 19-20 cmH2O) over a constant positive end-expiratory pressure (PEEP=1.5 cm H2O) were set to prevent atelectases. Post-synaptic neuromuscular blockade was induced on the first day (150 µg α-cobrotoxin) and maintained by continuous infusion (187 µg/day). Mechanical ventilation started after the neuromuscular blockade induction, avoiding hypercapnia and hypoxemia. Experiments were terminated after 5 days, 8 days, 10 days or 2-weeks. Female rats were used since diuresis was continuously monitored via an urethra catheter which is not trivial in a female rat but impossible in a male rat without introducing significant trauma (the experimental model has been modified to make it minimally invasive and the only skin incision is done in the neck region during catheterization of carotid artery and jugular vein). Diuresis was maintained above 1 ml/h. In no case did animals show any signs of infections or septicemia. The inspiratory air was humidified to reduce the risk of mucous formation in the respiratory tract and collected in condensing traps in the expiratory tract, frozen and stored at -20 0C and referred to as bronchoaveolar lavage (BAL) fluid.
Animals were euthanized under deep isoflurane anaesthesia by removal of the heart. Immediately after rats had been euthanized, the slow-twitch soleus, fast-twitch tibial anterior (TA), thoracic paravertebral, and diaphragm (mid-costal) muscles were gently dissected, immediately snap frozen in liquid propane chilled by liquid nitrogen and stored at -140 0C until further analysis.
Tissue preparation: Approximately 20-40 mg of frozen muscle tissue were quickly weighed in a 1.5 ml microtube (Tube 1). The tube was placed on ice and 10 volumes (200-400 µl) of ice-cold Buffer 1 (25 mM Tris, pH 7.2, 0.5% Triton X-100) was added. The muscle tissue was homogenized with a hand-held tight-fitting pestle and put back on ice until all samples were homogenized. The samples were centrifuged at 1500 g for 10 min at 40C. The supernatant (sarcoplasmic fraction) was pipetted off and placed into a new 1.5 ml microtube. The tubes were stored at -800C until further analysis. The pellet in Tube 1 was resuspended with 10 volumes (200-400 µl) of ice-cold Buffer 1 as a wash step. The tubes were centrifugated at 1500 g for 10 min at 40C, the supernatant pipetted off, and the pellet was put on ice to dry. Fifteen volumes of ice-cold Buffer 2 (20 mM Tris- HCl, pH 7.2, 100 mM KCl, 20 % Glycerol, 1 mM DTT,50 mM Spermidine) were added to the pellet in Tube 1, resuspended with a hand-held tight-fitting micro tube pestle; remaining/un-suspended protein is putatively collagen. The tube was centrifugated at 1500 g for 5 min at 4oC, and the supernatant (Myofibrillar fraction) was then transferred to a new tube and stored at -80oC until further analysis. Protein concentrations of sarcoplasmic isolates as well as putative myofibrillar resuspensions were quantified using the PierceTM BCA Protein Assay (Thermo Scientific) and read on a plate reader (Infinite M Nano+, Tecan) at an absorbance of 562 nm.
Myosin and actin protein measurements: Sarcoplasmic and myofibrillar fractions were diluted 1:10 in 8M Urea buffer (120 g urea, 38 g thiourea, 70 ml H2O, 25g mixed bed resin, 2,89g dithiothreitol, 1,51g Trizma base, 7,5g SDS), heated to 90 0C, centrifugated and loaded on a 12% sodium dodecyl sulphate- polyacrylamide gel electrophoresis (SDS-PAGE) for myosin and actin quantification. The acrylamide concentration was 4% (w/v) in the stacking gel and 12% in the running gel, and the gel matrix included 10% glycerol. Electrophoresis was performed at 16.0 mA for 5 h with a Tris–glycine electrode buffer (pH 8.3) at 15 0C (SE 600 vertical slab gel unit; Hoefer Scientific Instruments, San Francisco, CA, USA). The gels were Coomassie stained using SimplyBlue SafeStain (Invitrogen) and scanned in a gel scanner (GS-900 Calibrated Densitometer, Bio-Rad). The relative proportion of Myosin and Actin was determined using a densitometry system (Image Lab, Bio-rad).
Statistical analyses: One-way ANOVA was used to calculate differences between groups, and p<0.05 was considered statistically significant. Values are given as means±SD.
Results
There was no significant difference in the initial body weights between the different groups (see above), but there was a progressive decline in body weights with increasing duration of immobilization and mechanical ventilation (p<0.05-0.01) with a relative decline in body weight corresponding to 11±6%, 26±12%, 25±9%, and 33±9% in the 5-day, 8-day, 10-day, and 2-week groups, respectively. A corresponding decline in tibial anterior (TA) muscle weight was observed with a ~50% decline in muscle weight after 2 weeks of exposure to the ICU condition (immobilization and mechanical ventilation) compared with 0-day controls (601±57 mg), i.e., TA weights after 5 days, 8 days, 10 days, and 2 weeks were 481±89, 389±83, 393±191, and 289±67 mg, respectively.
Twice as much myosin as actin was observed in the control samples as expected giving an average myosin actin ratio of 2.1±0.1. A significant decline (p<0.001) in myosin vs. actin content was observed in all experimental groups compared with controls with myosin actin rations ranging between 1.1±0.3 and 1.2±0.3 with no significant differences in myosin actin ratios between the different groups exposed to immobilization and mechanical ventilation for 5 days and longer (Figure 1).
The relative myofibrillar vs. sarcoplasmic actin and myosin expression was determined in controls and the different groups exposed to immobilization and mechanical ventilation. The large majority of actin was detected in the myofibrliiar region and varying between 76±1.2 and 80±1.0 % between the different groups with no significant intergroup differences according to one-way ANOVA. No myosin was detected in the sarcoplasmic region and 100% myosin was detected in the myofibrillar preparation in all groups. All myosin detected were expressed in the myofibrillar preparation.
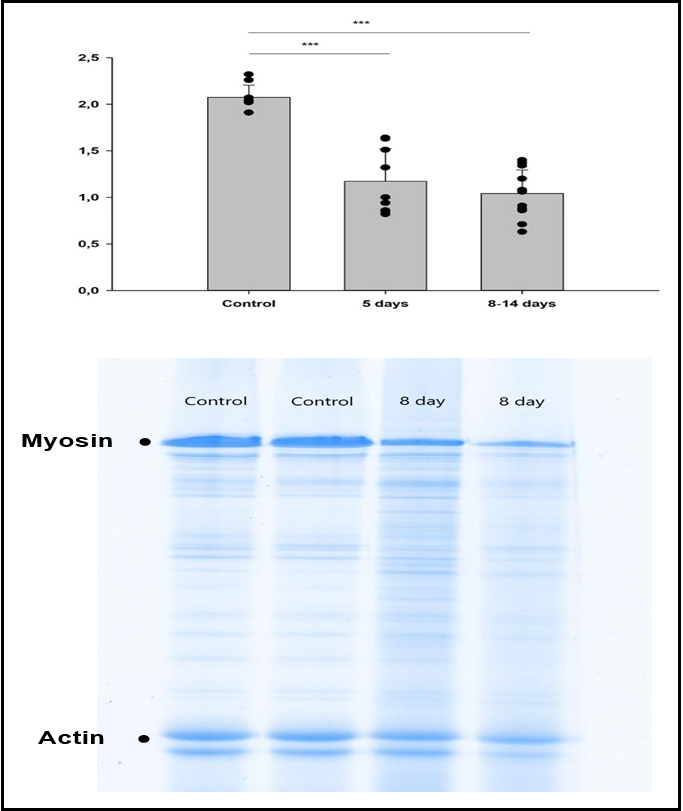
Figure 1: Electrophoretic separation of myofibrillar proteins and myosin actin ratios.
(A) Average myosin actin ratios measured on Coomassie stained 12% SDS-PAGE of tibial anterior muscle cross-sections in controls and rats exposed to 5 days and 8-14 days mechanical ventilation and immobilization. (B) Representative gels from two control rats (2.0 myosin actin ratio in both rats) and two rats exposed to 8 days mechanical ventilation and immobilization (myosin actin rations 0.9 and 0.6). *** denotes p<0.001.
Discussion
A preferential myosin loss represents the hallmark of CIM and we have used this as a diagnostic CIM criteria since we diagnosed the first patients with CIM in Scandinavia in 1995, initially using percutaneous muscle biopsies from leg muscles and more recently using a microbiopsy instrument which has the same diagnostic precision as the 10-20 fold larger percutaneous biopsies [4,7-9]. However, a redistribution of myosin and actin between the myofibrillar and sarcoplasmic compartments in skeletal muscle is a potential source of error in the diagnostic precision of the method, and it may also affect the recovery process during the weaning and post- ICU rehabilitation.
In accordance with previous studies, we see a preferential myosin loss in response to the ICU condition (immobilization and mechanical ventilation) using an established experimental ICU model. In our previous studies, a preferential loss of myosin was observed first after 5 days exposure to the ICU condition and progressing at longer durations in both the slow-twitch soleus and the fast-twitch extensor digitorum longus muscle [10,15]. In the current study, a ~50% lower myosin: actin ratio was observed after 5 days and remained at this level until the end of the two-week observation period, indicating a muscle-specific difference in the preferential myosin loss and an earlier decline in myosin content in the tibial anterior muscle than in soleus and extensor digitorum longus muscles. In spite of this, the relative distribution of myosin and actin in the myofibrillar and sarcoplasmic compartments did not differ from that of control animals.
In conclusion, the present results show a constant distribution of myosin and actin in the myofibrillar and sarcoplasmic compartment during the preferential myosin loss associated with critical illness myopathy, supporting its use as a diagnostic and prognostic marker.
Acknowledgements
This study was supported by grants from the Swedish Medical Research Council, Stockholm City Council (Alf 20150423, 20170133), ESICM, and VironMMI to LL
References
- Cacciani N, Skärlén Å, Wen Y, Zhang X, Addinsall AB, et al. (2022) A prospective clinical study on the mechanisms underlying critical illness myopathy-A time-course approach. Journal of cachexia, sarcopenia and muscle. 13(6): 2669-2682.
- Friedrich O, Reid MB, Van den Berghe G, Vanhorebeek I, Hermans G, et al. (2015) The Sick and the Weak: Neuropathies/Myopathies in the Critically Ill. Physiol Rev. 95(3): 1025-1109.
- Larsson L (2008) Acute quadriplegic myopathy: an acquired "myosinopathy". Adv Exp Med Biol. 642: 92-98.
- Larsson L, Li X, Edström L, Eriksson LI, Zackrisson H, et al. (2000) Acute quadriplegia and loss of muscle myosin in patients treated with nondepolarizing neuromuscular blocking agents and corticosteroids: mechanisms at the cellular and molecular levels [see comments]. Crit Care Med. 28(1): 34-45.
- Norman H, Zackrisson H, Hedstrom Y, Andersson P, Nordquist J, et al. (2009) Myofibrillar protein and gene expression in acute quadriplegic myopathy. J Neurol Sci. 285(1-2): 28-38.
- Samuelsson J, Zackrisson H, Tokics L, Ljungman P, Lidbrink E, et al. (1999) Acute quadriplegic myopathy following autologous peripheral blood stem cell transplantation for breast cancer. Bone Marrow Transplant. 23(8): 835-837.
- Marrero H, Stalberg EV, Cooray G, Corpeno Kalamgi R, Hedstrom Y, et al. (2020) Neurogenic vs. Myogenic Origin of Acquired Muscle Paralysis in Intensive Care Unit (ICU) Patients: Evaluation of Different Diagnostic Methods. Diagnostics (Basel). 10(11): 966.
- Larsson L, Roland A (1996) [Drug induced tetraparesis and loss of myosin. Mild types are probably overlooked]. Lakartidningen. 93(23): 2249-2254.
- Cacciani N, Skärlén Å, Wen Y, Zhang X, Addinsall AB, et al. (2022) A prospective clinical study on the mechanisms underlying Critical Illness Myopathy – A time-course approach. Journal of cachexia, sarcopenia and muscle. 13(6): 2669-2682.
- Ochala J, Gustafson AM, Diez ML, Renaud G, Li M, et al. (2011) Preferential skeletal muscle myosin loss in response to mechanical silencing in a novel rat intensive care unit model: underlying mechanisms. J Physiol. 589(Pt 8): 2007-2026.
- Corpeno Kalamgi R, Salah H, Gastaldello S, Martinez-Redondo V, et al. (2016) Mechano-signalling pathways in an experimental intensive critical illness myopathy model. J Physiol. 594(15): 4371-4388.
- Roberts MD, Young KC, Fox CD, Vann CG, Roberson PA, et al. (2020) An optimized procedure for isolation of rodent and human skeletal muscle sarcoplasmic and myofibrillar proteins. J Biol Methods. 7(1): e127.
- Dworkin BR, Dworkin S (1990) Learning of physiological responses: I. Habituation, sensitization, and classical conditioning. Behav Neurosci. 104(2): 298-319.
- Dworkin BR, Dworkin S (2004) Baroreflexes of the rat. III. Open- loop gain and electroencephalographic arousal. Am J Physiol Regul Integr Comp Physiol. 286(3): R597-605.
- Norman H, Nordquist J, Andersson P, Ansved T, Tang X, et al. (2006) Impact of post-synaptic block of neuromuscular transmission, muscle unloading and mechanical ventilation on skeletal muscle protein and mRNA expression. Pflugers Arch. 453(1): 53-66.