


Mahmoud Abu Zahra1*, Haitham Al-Dhmour1, Neha Patel1, Jacqueline Weingarten-Arams2, Samuel Gorstein2, Henry M. Ushay2, Joshua Hanau3, Jenny Shliozberg3, Danielle Weiss4, Michelle R. Ewart5, Aneela Bidiwala1
1Division of Pediatric Respiratory and Sleep Medicine, The Children’s Hospital at Montefiore, Albert Einstein College of Medicine, Bronx, NY. ORCID ID: 0000-0002-9682-0569
2Division of Pediatric Critical Care Medicine, The Children’s Hospital at Montefiore, Albert Einstein College of Medicine, Bronx, NY.
3Divison of Allergy and Immunology, The Children’s Hospital at Montefiore, Albert Einstein College of Medicine, Bronx, NY.
4Division of Pediatric Radiology, The Children’s Hospital at Montefiore, Albert Einstein College of Medicine, Bronx, NY.
5Department of Pathology, The Children’s Hospital at Montefiore, Albert Einstein College of Medicine, Bronx, NY.
*Corresponding Author: Mahmoud Abu Zahra, Division of Pediatric Respiratory and Sleep Medicine, The Children’s Hospital at Montefiore, Albert Einstein College of Medicine, Bronx, NY. ORCID ID: 0000-0002-9682-0569.
Abstract
Children’s interstitial lung disease (chILD) is a heterogeneous group of disorders that encompasses genetic, infectious and inflammatory diseases, presenting with respiratory symptoms and diffuse abnormalities on lung imaging. It is a rare entity with wide differential diagnosis and overlapping presentations, causing a diagnostic conundrum for physicians. When evaluating a patient with chILD syndrome, it is generally recommended to rule out common diseases first, including infections, immunodeficiency, structural airway abnormalities, and congenital heart disease, but a comprehensive work up might be warranted in rare diseases. We describe a rare case of an infant who presented with diffuse lung involvement in the setting of CGD, a diagnosis which was challenging and missed on the initial immunologic evaluation.
Keywords: chILD, Interstitial lung disease, Chronic granulomatous disease, Immunodeficiency, lung imaging, genetic.
Introduction
Infants with diffuse lung disease are considered to have chILD syndrome if they meet three out of the following criteria: 1- respiratory symptoms (cough, rapid breathing), 2- respiratory signs (tachypnea, retractions, adventitious sounds), 3- hypoxemia, and 4- diffuse abnormalities on lung imaging (chest x-ray or computed tomography) [1]. It is a non-specific terminology which is used when a definitive diagnosis has not yet been established [1,2]. A step wise approach is advised when evaluating these cases, which focuses on history, physical examination findings, and the use of investigations while minimizing risks. [1,2]. We report a case of an infant who presented significant diagnostic and management challenges and ultimately was diagnosed with chronic granulomatous disease (CGD).
Case Report
A male infant born prematurely at 34 4/7 weeks via cesarean section for severe preeclampsia, had a 27-day neonatal intensive care unit (NICU) course during which he required continuous positive airway pressure (CPAP) for 1 week, then was weaned off all respiratory support. However, he continued to have intermittent tachypnea which resolved with a single furosemide dose. Chest x-rays showed sub-segmental atelectasis, echocardiography was normal, and he was discharged home on room air.
At home, he developed worsening cough, fever, and tachypnea leading to emergency department presentation at four months of age. On physical exam, lungs were clear to auscultation, oxygen saturation was 89% and no clubbing was appreciated. Chest x-ray showed bilateral hazy and patchy opacities [figure1]. Blood work demonstrated an elevated white blood cells count and respiratory viral panel was negative. He was admitted to the pediatric intensive care unit (PICU) and treated with high flow nasal cannula 8 LPM FiO2 40% and antibiotics.
His course was notable for worsening tachypnea and poor weight gain despite high calorie diet. Arterial blood gas showed adequate ventilation and minimal alveolar-arterial gradient. Respiratory support escalated to non-invasive neurally adjusted ventilatory assist (NAVA). Chest CT was completed and showed bilateral diffuse hazy ground glass opacities with lower lobe predominance [figure1]. Lymphocyte subset analysis and quantitative immunoglobulins were within normal limits. Pulse steroid therapy, hydroxychloroquine, and anti-inflammatory dose azithromycin were started for management of presumed chILD. Genetic testing for ILD (Invitae panel (3)) was sent.
Flexible bronchoscopy revealed normal anatomy and thoracoscopic lung biopsy was performed at the same time. Bronchoalveolar lavage (BAL) grew Pseudomonas aeruginosa. Genetic testing for ILD (Invitae panel (3)) was negative. Lung biopsy did not reveal alveolar structure or surfactant protein abnormality but showed evidence of pneumonia with chronic remodeling. Cefepime and alternating monthly nebulized Tobramycin were started for the treatment of Pseudomonas.
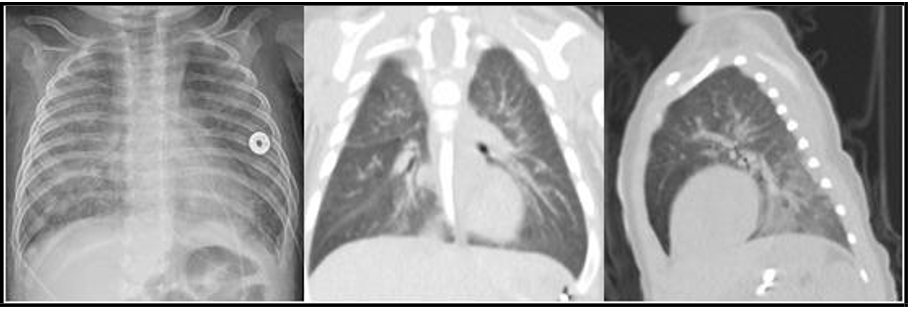
Figure 1: CXR (left) showing bilateral hazy and patchy opacities, Chest CT (middle and right) coronal and sagittal views showing ground glass opacities with lower lobe predominance, at the age of 4 months
Given an atypical organism cultured from BAL, an oxidative burst assay was performed (Dihydrorhodamine (DHR)) which revealed absent neutrophil function. Whole exome sequencing (WES) detected pathogenic variant in CYBB gene (c.141+5G>C, Intronic, Hemizygous) confirming the diagnosis of CGD. Diagnostic work-up is summarized in [figure2].
Hence, prophylaxis with sulfamethoxazole-trimethoprim and itraconazole was started. Nevertheless, the baby continued to have poor weight gain and persistent tachypnea, ultimately requiring a tracheostomy.
He continues to be tracheostomy and ventilator dependent. He developed recurrent pneumonias with subsequent CT imaging showing disease progression and involvement of upper and middle lobes [figure3]. He was started on Interferon gamma therapy at the age of 11 months, however, he has continued to experience recurrent lung infections.
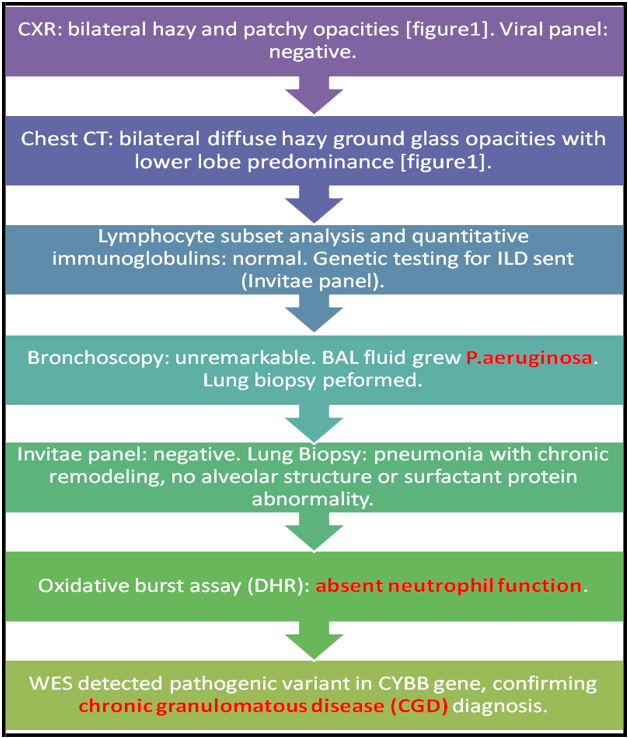
Figure 2: Summary of diagnostic work-up
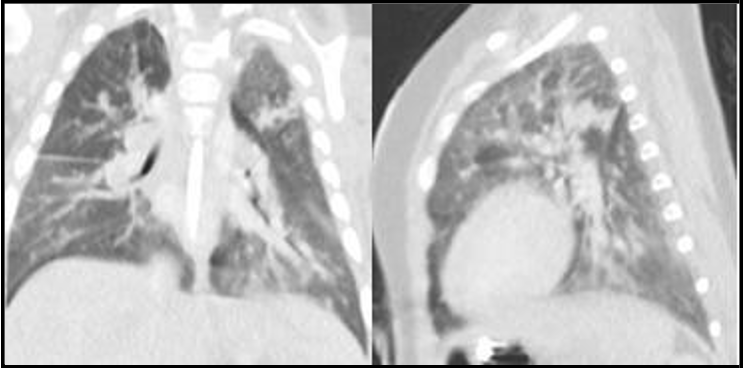
Figure 3: Chest CT coronal and sagittal views showing disease progression and involvement of upper and middle lobes, at the age of 7 months
Discussion
chILD comprises a diverse group of disorders classified together due to similar clinical, radiographic, or pathological manifestations [1,4]. Recent approaches tend to divide these entities based on the age at presentation to disorders more prevalent in infancy and disorders not specific to infancy [4,5]. Within this classification, it is also important to address different histopathological patterns, and the underlying etiology, whether it is a primary pulmonary process or a systemic disorder with pulmonary involvement [1,5-7].
These classifications are meant to guide physicians to help facilitate diagnostic evaluation and treatment plans. However, establishing a diagnosis is not always straight forward, thus, a diagnostic algorithm was suggested by the American Thoracic Society in 2013 which focused on ruling out underlying diseases first, including infections, congenital heart disease, immunodeficiency, cystic fibrosis, and aspiration [1]. It also emphasized the role of family history of chILD in narrowing the differential, indications for genetic testing, chest CT scan findings, and bronchoscopy with BAL, which could reveal the final diagnosis without the need for lung biopsy [1]. Nevertheless, lung biopsy remains the mean of definitive diagnosis for many forms of chILD in which the initial evaluation is inconclusive [1,8].
It is worth mentioning that in clinical practice, multiple factors play a role in determining the urgency and the choice of the diagnostic tests. Decisions are often made while taking into account disease severity, progression, duration, and the patient’s overall clinical status. In our case, the patient underwent extensive work-up and lung biopsy due to worsening clinical condition and an indecisive initial work up.
Treatment of chILD focuses on supportive therapy with oxygen and ventilatory support, adequate caloric intake, and interventions tailored to the specific etiology [1,6]. Examples of specific interventions include antimicrobials for infections, management of swallowing dysfunction in aspiration syndrome, and whole lung lavage in patients with alveolar proteinosis [1,9]. Empirical pharmacological therapy is often considered, including systemic steroids, which proves beneficial in cases where inflammation and inappropriate cellular proliferation play a role [10], hydroxychloroquine, which was reported to have beneficial effects in some cases with SFTPC and ABCA3 mutations, and azithromycin due to its anti-inflammatory and immunomodulatory effects [6,11,12]. We believe that treatment decisions should be tailored based on the individual’s presentation, risk factors, and definitive etiology (if established), while minimizing medications side effects. In our case, a decision was made to start empirical treatment due to rapid clinical deterioration initially, and once a final diagnosis was made, CGD related therapies were started.
Our patient had a presentation consistent with chILD but it was difficult to determine the etiology. Suspicion for immunodeficiency arose when BAL grew Pseudomonas aeruginosa, a catalase positive organism [13], that does not commonly cause pneumonia in healthy subjects within this age group. Then, CGD diagnosis was established by DHR and WES.
CGD is a rare inherited immunodeficiency caused by loss-of-function variants of genes encoding subunits of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase complex (14, 15). Hemizygous variants in the CYBB gene cause X-linked disease, which is the most common mode of inheritance. Other mutations like CYBA, CYBC1, NCF1, NCF2, and NCF4 were reported to cause autosomal recessive disease [15]. Loss of NADPH function leads to impaired phagocytic killing of organisms with the inability to produce reactive oxygen species (ROS) in the phagosome [14,15].
Diagnosis of CGD is based on evaluating ROS production by neutrophils. DHR is now considered the gold standard for diagnosis [16]. In this test, neutrophils are stimulated with phorbol myristate acetate then incubated with DHR 123, which is oxidized to rhodamine 123 by the normal NADPH oxidase-myelopeoxidase activity in neutrophils. Rhodamine 123 emits fluorescence which is assessed by flow cytometry as a surrogate measure of NADPH oxidase function [16,17]. The test has high sensitivity and provides reliable quantitative results, but genetic testing is often performed to identify the mutation and inform prognosis [16].
Patients with this disease are prone to recurrent infections with catalase positive organisms, such as Staphylococcus aureus, Burkholderia cepacia, Serratia, Aspergillus, Nocardia, and Pseudomonas aeruginosa (18), although the latter is not commonly reported which adds to the uniqueness of our case [18]. Infections commonly involve the lungs, skin, lymph nodes and liver. Secondary interstitial lung disease often develops later in life due to recurrent pneumonia and inflammation [14,15].
Managing CGD starts with antimicrobial, antifungal, and interferon gamma prophylaxis, which demonstrated improved overall survival [14]. However, hematopoietic stem cell transplantation remains the only curative treatment [14], an option that was not strongly considered in this patient due to his complicated course with recurrent infections and significant lung disease.
We believe our case was atypical and unique, the patient acquired pseudomonas early in his life, his disease progressed rapidly leading to secondary ILD, and the severity was significant, resulting in the need for persistent non-invasive support, and eventually tracheostomy with chronic mechanical ventilation. Data published from registries in the US and Europe in 2000 and 2009, reported mean age for CGD diagnosis of 3 years and 4.9 years for X-linked disease, and 7.8 years and 8.8 years for autosomal recessive disease, respectively [19,20]. Later publications reported earlier neonatal manifestations of CGD, which included pulmonary aspergillosis, skin infections, osteomyelitis, and pneumonia [15], however, no infant has been reported to have ILD due to CGD prior to this case.
Conclusion
Children with diffuse lung disease present a diagnostic dilemma for physicians due to overlapping presentations and imaging findings. Nevertheless, establishing a definitive diagnosis is vital to inform prognosis, genetic counselling, and to determine treatment choices. A stepwise approach is recommended, in which common entities are ruled out first, including infections and immunodeficiency. Extensive work up is warranted in some cases with negative initial evaluation, and multidisciplinary teamwork collaboration typically proves beneficial. CGD is a rare immunodeficiency which could lead to secondary ILD and should be suspected in the presence of catalase positive infectious etiology.
Statements and Declarations:
Funding: No funding was secured for this case report.
Competing interests: The authors have no relevant financial or non-financial interests to disclose.
Author Contributions: All authors contributed to patient care. The first draft of the manuscript was written by Mahmoud Abu Zahra1, and all authors commented on previous versions of the manuscript. All authors read and approved the final manuscript.
Acknowledgments: None to declare.
References
- Kurland G, Deterding RR, Hagood JS, Young LR, Brody AS, et al. (2013) An official American Thoracic Society clinical practice guideline: classification, evaluation, and management of childhood interstitial lung disease in infancy. Am J Respir Crit Care Med. 188(3): 376-94.
- Vece TJ, Young LR (2016) Update on Diffuse Lung Disease in Children. Chest. 149(3): 836-45.
- Invitae Neonatal Respiratory Distress Panel.
- Deutsch GH, Young LR, Deterding RR, Fan LL, Dell SD, et al. (2007) Diffuse lung disease in young children: application of a novel classification scheme. Am J Respir Crit Care Med. 176(11): 1120-8.
- Fan LL, Dishop MK, Galambos C, Askin FB, White FV, et al. (2015) Diffuse Lung Disease in Biopsied Children 2 to 18 Years of Age. Application of the chILD Classification Scheme. Ann Am Thorac Soc. 12(10): 1498-505.
- Ferraro VA, Zanconato S, Zamunaro A, Carraro S (2020) Children's Interstitial and Diffuse Lung Diseases (ChILD) in 2020. Children (Basel). 7(12): 280.
- Liang T, Vargas SO, Lee EY (2019) Childhood Interstitial (Diffuse) Lung Disease: Pattern Recognition Approach to Diagnosis in Infants. AJR Am J Roentgenol. 212(5): 958-67.
- Chan CD, Niyogi A, Jaffray B, Brodlie M, Gabra H (2021) Lung biopsy in children: when is it useful? Arch Dis Child. 106(3): 291- 293.
- Bush A, Pabary R (2020) Pulmonary alveolarproteinosis in children. Breathe (Sheff). 16(2): 200001.
- Bush A, Cunningham S, de Blic J, Barbato A, Clement A, et al. (2015) European protocols for the diagnosis and initial treatment of interstitial lung disease in children. Thorax. 70(11): 1078-84.
- Avital A, Hevroni A, Godfrey S, Cohen S, Maayan C, et al. (2014) Natural history of five children with surfactant protein C mutations and interstitial lung disease. Pediatr Pulmonol. 49(11): 1097-105.
- Williamson M, Wallis C (2014) Ten-year follow up of hydroxychloroquine treatment for ABCA3 deficiency. Pediatr Pulmonol. 49(3): 299-301.
- Elkins JG, Hassett DJ, Stewart PS, Schweizer HP, McDermott TR (1999) Protective role of catalase in Pseudomonas aeruginosa biofilm resistance to hydrogen peroxide. Appl Environ Microbiol. 65(10): 4594-600.
- Yu HH, Yang YH, Chiang BL (2021) Chronic Granulomatous Disease: a Comprehensive Review. Clin Rev Allergy Immunol. 61(2): 101-113.
- Marzollo A, Conti F, Rossini L, Rivalta B, Leonardi L, et al. (2022) Neonatal Manifestations of Chronic Granulomatous Disease: MAS/HLH and Necrotizing Pneumonia as Unusual Phenotypes and Review of the Literature. J Clin Immunol. 42(2): 299-311.
- Kuhns DB (2019) Diagnostic Testing for Chronic Granulomatous Disease. Methods Mol Biol. 1982: 543-571.
- Yu JE, Azar AE, Chong HJ, Jongco AM, Prince BT (2018) Considerations in the Diagnosis of Chronic Granulomatous Disease. J Pediatric Infect Dis Soc. 7(suppl_1): S6-s11.
- Marciano BE, Spalding C, Fitzgerald A, Mann D, Brown T, et al. (2015) Common severe infections in chronic granulomatous disease. Clin Infect Dis. 60(8): 1176-83.
- Winkelstein JA, Marino MC, Johnston RB, Boyle J, Curnutte J, et al. (2000) Chronic granulomatous disease. Report on a national registry of 368 patients. Medicine (Baltimore). 79(3): 155-69.
- van den Berg JM, van Koppen E, Ahlin A, Belohradsky BH, Bernatowska E, et al. (2009) Chronic granulomatous disease: the European experience. PLoS One. 4(4): e5234.