


N. Ramlachan1,2*, W. Ramcharan3, R. Ramcharan3
1Genix Diagnostics Limited, Champs Fleurs, Trinidad
2University of Trinidad and Tobago, Tamana, Trinidad
3Department of Neurosurgery, Department of Clinical Surgical Sciences, Port of Spain General Hospital, University of the West Indies, Trinidad and Tobago
*Corresponding Author: N. Ramlachan, Genix Diagnostics Limited, Champs Fleurs, Trinidad, University of Trinidad and Tobago, Tamana, Trinidad.
Abstract
Objective: To present a case of both Lynch and Von Hippel Lindau Syndromes diagnosed in a 38-year-old male patient, confirmed by genetic analyses, who presented with the obstructive hydrocephalus secondary to multiple posterior fossa hemangioblastomas and benign pancreatic cysts.
Designs and Methods: Single case report from a neurosurgery facility at the above institution. Genetic testing was performed off-site to confirm the hereditary cancer genotypic profile.
Results: This patient was treated via an Endoscopic Third Ventriculostomy for the obstructive hydrocephalus. Genetic testing detected a pathogenic variant, PMS2, Deletion (Exons 13-14), associated with Lynch syndrome and another pathogenic variant, VHL, Deletion (Entire Sequence), consistent with von Hippel-Lindau syndrome. The patient was consequently diagnosed using genetic testing, with Von Hippel Lindau syndrome, a rare familial multi-organ autosomal dominant tumour syndrome characterized by the presence of by visceral cysts and benign tumours in multiple organ systems seen in 1 in 32,000 live births. Patient was also diagnosed incidentally with Lynch syndrome, with a deleterious mismatch repair PMS2 gene mutation; seen in 1 in every 370 patients and in 2-3% of colorectal carcinomas. He is currently under surveillance for additional associated tumours.
Conclusion: The case study reports an unusual case of both Lynch syndrome and Von Hippel Lindau syndrome in a 38-year-old male patient, thereby underscores the necessity for screening of suspected genetic diseases in order to facilitate the earlier detection and treatment of associated tumors in familial cancers.
Case Presentation
A 39-year-old East Indian male, with no known co-morbidities presented with a history of global generalized headaches, early in the morning nausea and vomiting with a progressive decline in his coordination and gait for 2 months. On examination, normal vital signs were recorded, and neurological examination was grossly normal, except for incoordination, the left was more than the right.
He had a complete blood count and renal function tests, which were normal. His radiological investigations- CT Scan Brain was reported as multiple dural based lesions within the posterior fossa, likely metastases, which effacement of the 4th ventricle with resultant obstructive hydrocephalus.
A whole-body CT-Scan was performed which only positive finding were multiple simple cysts throughout the pancreas of multiple hemangioblastomas within both cerebellar hemispheres (Figure 1,2 & 3).
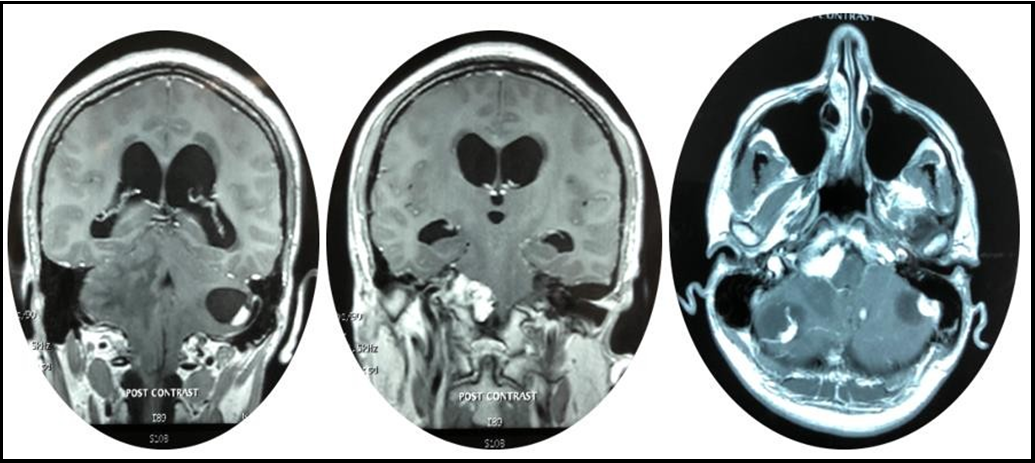
Figures 1,2 & 3: demonstrating axial and coronal slices of MRI brain showing multiple lesions within the posterior fossa (Blue arrowhead) with both white and grey matter edema (Red arrowhead) and hydrocephalus via dilated temporal horns of bilateral lateral ventricles (Purple arrow) and periventricular edema (Green arrow).
The patient was started on high dose corticosteroids and consented for an Endoscopic Third Ventriculostomy. Endoscopic Third Ventriculostomy was performed via Right Kocher’s Burr Hole and Rigid endoscopy. The frontal horn of the right ventricle was entered, and the Foramen of Munroe was identified. The third ventricle was the entered via the Right Foramen of Munroe and the ventriculostomy was performed via blunt dissection through the tuber cinereum into the pre-pontine cistem. The patient recovered well from the surgery and was referred to Genix Diagnostics Ltd., for genetic testing to investigate any possible association with Von Hippel Lindau Syndrome.
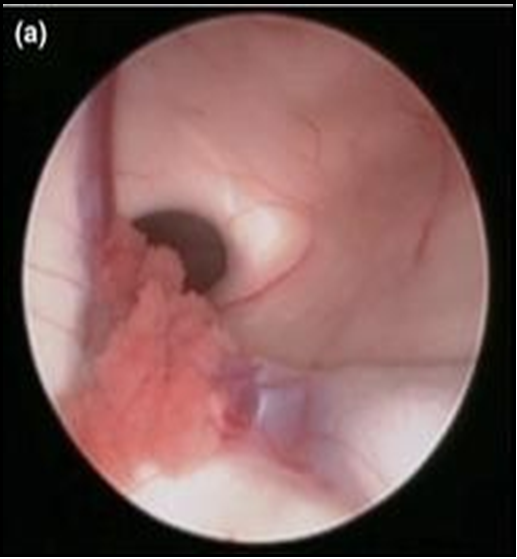
Figure 4: Endoscopic view of Right Frontal Horn of lateral Ventricle showing:
A: Foramen of Munroe
B: Choroid Plexus
C: Thalamostriate vein D: Anterior Septal vein
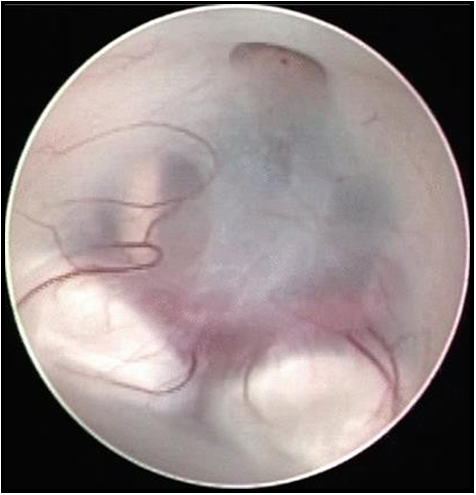
Figure 5: Endoscopic view of anterior floor of third ventricle showing:
A: Infundibular recess
B: Tuber Cinereum
C: Posterior Cerebral Arteries
D: Mammillary Bodies’
E: Left Occulomotor nerve
F: Hypothalamus
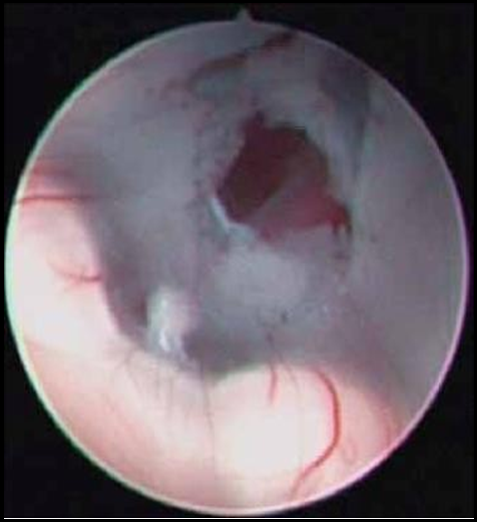
Figure 6: Endoscopic view of fenestrated third ventricular floor
A: Fenestration through tuber cinereum
B: Infundibular recess
C: Mammillary Bodies
Genomic analysis of DNA obtained from saliva samples was enriched for targeted regions using a hybridization protocol and sequenced using Illumina technology. All targeted regions were sequenced with more than 50X depth and reads aligned to a reference, which was interpreted in the context of a clinically relevant transcript. Sequence analysis identified pathogenic variant, PMS2 Deletion (Exons 13-14) using deletion sequencing in reference to Thompson et al 2013 [10]; and pathogenic variant VHL, Deletion (Entire Sequence) according to the methodology proposed by Aarnio et al 2000 [2].
Genomic analyses therefore allowed for the diagnosis of both von Hippel-Lindau Syndrome and Lynch Syndrome (also called hereditary nonpolyposis colorectal cancer (HNPCC) syndrome) in this individual.
The Patient was then started on a surveillance program for Von Hippel Lindau Syndrome, which included.
Annual:
1. Dilated Retinal Exams
2. Full body Physical examinations
3. Blood and urinary 24-hour cathecoamines and metanephrins
4. CT abdomen and pelvis Scans
Two yearly:
1. MRI Scans of the brain and Spinal Cord
2. Complete Audiology Examination
Surveillance for Lynch Syndrome:
1. Colonoscopy every 2-3 years, until age 50, then every 1-2 years after 50.
Thus far, these surveillance tests have been negativing for associated tumors.
Discussion
Genetic analysis revealed a pathogenic variant that results in a “loss of function” in the PMS2-gene associated with autosomal dominant Lynch (or HNPCC) syndrome, characterized by an increased risk of colorectal cancer, as well as cancers of the stomach, small intestines, hepatobiliary tract, pancreas, urinary tract and brain [3]. Women with pathogenic variants in PMS2 have a 15-20% risk of colorectal cancer and a 15% risk of endometrial cancer [3]. The variant identified, PMS2 Deletion (Exons 13-14), creates a premature translational stop signal and is expected to result in an absent or disrupted protein product [8]. This deletion eliminates a portion of the MLH1 interaction domain, which is required for normal protein function [8]. This specific variant has not been reported in the literature in individuals with PMS2-related disease.
We also identified a pathogenic variant, VHL, Deletion (Entire Sequence) associated with autosomal dominant von Hippel-Lindau (VHL) syndrome and autosomal recessive familial erythrocytosis, type 2 [9]. A gross deletion of the genomic region encompassing the full coding sequence of the VHL gene, was detected in this individual. This result has been reported in multiple individuals and families with VHL, and is consistent with a predisposition to, or diagnosis of, VHL- related conditions [2]. VHL is characterized by the development of cysts and tumours, most of which are benign, however individuals with VHL have a 40-70% risk of developing clear cell renal carcinoma and an 8-17% risk of developing pancreatic neuroendocrine tumours [9]. Other associated tumour types include hemangioblastomas of the CNS and retina, pheochromocytomas, endolymphatic sac tumours, epididymal cystadenomas and head and neck paragangliomas [10]. Patient follow-up for VHL and Lynch Syndromes and Familial testing for these variants were both recommended but declined to date by the family.
Early diagnosis and treatment of VHL and LS is possible and recommended for families with positive pathogenic mutations deemed causative in at least on proband. For example, laser treatment for retinal hemangioblastomas at an asymptomatic stage, may affect prognosis of VHL patients positively. Therefore, it is important to know your genetic risk and undergo life long, regular surveillance in these familial hereditary cancers. Also, high risk VHL or LS family members who are either symptomatic carriers or are carriers for mutation are advised to undergo periodic multidisciplinary surveillance according to published international guidelines. The surveillance guidelines for LS or VHS combine clinical, biochemical and radiological investigations, an annual physical examination (e.g. colonosocopies or opthamological investigations, respectively) during lifetime. For instance, this can start as early as 5 years of age with retinal examinations for VHL family members with known pathogenic mutations [11].
The substantial decrease in morbidity and mortality of many familial cancers has been largely attributed to the implementation of periodic surveillance (most often due to confirmed genetic risk) in addition to surgical technique advances. Hence, the importance of genomic analyses coupled with increased surveillance to diagnose, treat, and improve the prognosis of VHL and LS and other familial hereditary cancer syndromes. Emphasis on genetic counseling and further familial analyses must be encouraged by physicians to avoid the development of familial cohorts of syndromic cancers only symptomatically diagnosed.
Conclusion
Genetic analysis proved beneficial for the diagnosis of this patient, who presented with multiple obstructive hydrocephalus secondary to multiple posterior fossa hemangioblastomas and benign pancreatic cysts. This shows the significance of genetic analysis as a diagnostic tool for the early detection, predisposition to, and diagnosis of diseases. This case also reveals the need for familial testing within this family to determine risks of VHL-related diseases and, Lynch syndrome, and other hereditary cancers, especially those that are syndromic and can improve prognostics with early surveillance and intervention. Education and further exposure to the utility of genetic testing is recommended for both patients and physicians in public and private health facilities in Trinidad and Tobago and regionally.
References
- Meier PM, Guzman R, Erb TO (2014) Endoscopic paediatric neurosurgery: Implications for anaesthesia. Journal of Paediatric Anaesthesia. 24(7): 668-77.
- Hes F, Zewald R, Peeters T, Sijmons R, Links T, et al. (2000) Genotype-phenotype correlations in families with deletions in the von Hippel-Lindau (VHL) gene. Human Genetics 106(4): 425-31.
- Aarnio M, Mecklin JP, Aaltonen LA, Nyström-Lahti M, Järvinen HJ (1995) Life‐time risk of different cancers in hereditary non‐polyposis colorectal cancer (HNPCC) syndrome. International Journal of Cancer. 64(6): 430-433.
- Sengul G, Tuzun Y, Cakir M, Duman S, Colak A, et al. (2012) Neuroendoscopic Approach to Quadrigeminal Cistern Arachnoid Cysts. Eurasian Journal of Medicine. 44(1): 18-21.
- Handbook of Neurosurgery, 8th Edition by Greenburg et al, January 2010.
- NCBI – MedGen database, dbSNP database
- NCCN Surveillance guidelines for Von Hippel Lindau and Lynch Syndromes.
- Guerrette S, Acharya S, Fishel R (1999) The interaction of the human MutL homologues in hereditary nonpolyposis colon cancer by Guerette, Acharya and Fishel. Journal of Biological Chemistry. 274(10): 6336-6341.
- van Leeuwaarde RS, Ahmad S, van Nesselrooij B, Zandee W, Giles RH (2015) Von Hippel-Lindau Syndrome. Gene Reviews.
- Thompson BA, Spurdle AB, Plazzer JP, Greenblatt MS, Akagi K, et al. (2014) Application of a 5-tiered scheme for standardized classification of 2,360 unique mismatch repair gene variants in the InSiGHT locus-specific database by Thompson at al. Nature Genetics. 46(2): 107-115.
- Poulsen ML, Budtz-Jorgensen E, Bisgaard ML (2010) Surveillance in von Hippel-Lindau disease (vHL). Clin Genet. 77(1): 49-59.