


Khadija M. Alshehabi¹*, Sumayah Askandarani²
1Nephrology, Salmaniya Medical Complex, Government Hospitals, Manama, Bahrain
2Nephrology, King Fahad Specialist Hospital, Eastern Health Cluster, Dammam, Saudi Arabia
*Corresponding Author: Khadija M. Alshehabi, Nephrology, Salmaniya Medical Complex, Government Hospitals, Manama, Bahrain.
Abstract
Fabry disease is a rare X-linked lysosomal storage disorder due to mutations in the GLA gene causing the deficiency of α-galactosidase A (α- gal A). Several systems are involved in this disease, including the kidneys leading to End Stage Kidney Disease. We now report a case series for ten family members screened and tested positive for Fabry disease after evaluating the index case post-kidney transplantation.
Keywords: Fabry Disease, Enzyme Replacement Therapy, End Stage Kidney Disease, Cardiomyopathy, Post-Kidney Transplantation
Introduction
Fabry disease is a rare progressive multisystem X-linked lysosomal storage disorder caused by mutations in the GLA gene leading to α- galactosidase A (α-gal A) deficiency. This enzyme is a lysosomal enzyme involved in glycosphingolipid catabolism [1]. Reduced activity or absence of α-galactosidase A (α-gal A) enzyme results in progressive accumulation of glycolipids, mainly globotriaosylceramide (GL-3, Gb3) and its derivative, globotriaosylsphingosine (also-GL-3), in plasma and a wide range of cells in the body such as vascular endothelial cells, cardiomyocytes, arterial smooth muscle cells, and other cell types in the kidneys and the nervous system [2]. Fabry disease is found in all races and regions worldwide, with an estimated incidence of 1:40,000 – 1:170,000 in the general population [3]. In the Gulf countries, the prevalence of Fabry disease in the United Arab Emirates (UAE), for example, was estimated to be around 0.25 per 100,000 live births [4], while Al- Sannaa NA et al.reported the prevalence of Fabry disease in the Eastern province of Saudi Arabia to be around 42.2 per 100,000 live births [5]. A prospective study by Salwa et al. recently assessed the prevalence of Fabry disease among Saudi patients on hemodialysis in 3 major hospitals. The majority of Fabry disease in this cohort was 4.8 per 1000 patients [6].
At present, more than 1000 GLA gene variants have been discovered [3], and this may be the cause for its broad spectrum of clinical phenotypes ranging from “classic” severe form in males to asymptomatic females [7]. Classical Fabry disease may present initially with chronic neuropathic pain and episodic painful pain crises that emerge during childhood and then involve other systems manifested by eyes, skin abnormalities, gastrointestinal (GI) disturbances, kidney injuries, and further complications in the heart, lung, and central nervous system [2]. Heterozygotes may present late and display all disease symptoms, including pain, orthostatic hypotension, angiokeratoma, ocular abnormalities, cochleovestibular involvement, gastrointestinal symptoms, and respiratory involvement [8].
In this context, we report ten cases from one family in which they tested positive for Fabry disease during screening by enzyme assay. Some developed complications, such as cardiac disease and End Stage Kidney Disease (ESKD) and are being monitored for disease manifestations. All of them were referred for enzyme replacement therapy.
Case Reports
Case 1: index
Mr. S is a 43-year-old male from the AL-Hassa governorate in the Eastern Province of Saudi Arabia, who presented to our transplant center in 2018 with ESKD with unknown etiology. He required hemodialysis for almost two years, and then he received a living-related kidney transplant from his bother in October 2019 with excellent immediate graft function. The patient’s immunosuppression regimen comprised intravenous anti-thymocyte globulin and triple immunosuppression (prednisone, tacrolimus, and mycophenolate mofetil). His post-transplant course was uneventful, with serum creatinine around 0.8 to 1 mg/dl without significant proteinuria. However, two months later, he reported progressive generalized body fatigue and chronic constipation, in addition to the development of punctate skin lesions on his arms, which warranted a skin biopsy.
The skin biopsy showed sections of epidermal hyperplasia characterized by acanthosis, elongation of rete ridges, and hyperkeratosis with the epidermis encircling the dilated vascular space. Often thrombosis within the vascular ectasia was also present (Figure 1). According to the skin biopsy results, additional investigations were requested. The patient was evaluated for α- galactosidase A (α-Gal A) enzyme activity which came to be markedly deficient in the recipient (0.08 nmol /hour /mL in plasm) (Figure 2), and diagnosis of Fabry disease (FD) was confirmed. Further evaluation of his respiratory, gastrointestinal, central nervous (CNS), and cardiovascular systems were unremarkable. The kidney function in his respective donor remained normal (serum creatinine around 1 mg/dl with no proteinuria), and his alpha-galactosidase level was normal.
The Family history showed that the recipient’s parents, who were close relatives, died at a young age with unclear cause. The index case has four brothers with no sisters. Two of his brothers were healthy, while the other two had ESKD of unknown etiology (Figure 3). The alpha-galactosidase enzyme activity was measured for the daughter of the index case and his affected brothers. The recipient was referred to a local metabolic center to initiate Enzyme Replacement Therapy (ERT).
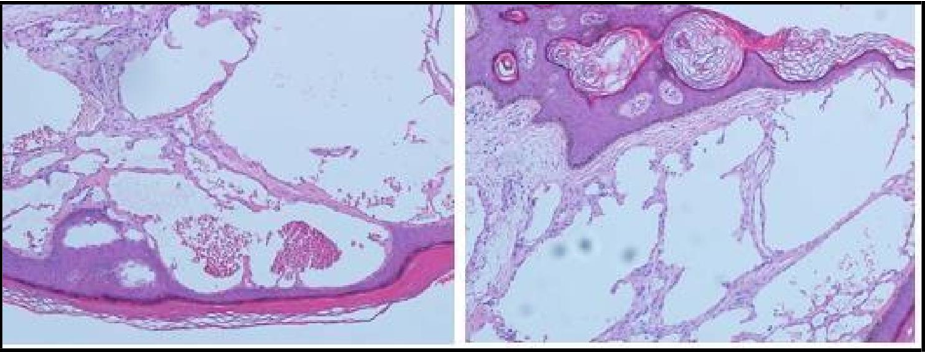
Figure 1: Skin Biopsy: Epidermal hyperplasia and thrombosis within the vascular ectasia

Figure 2: Genetic testing confirms Fabry disease
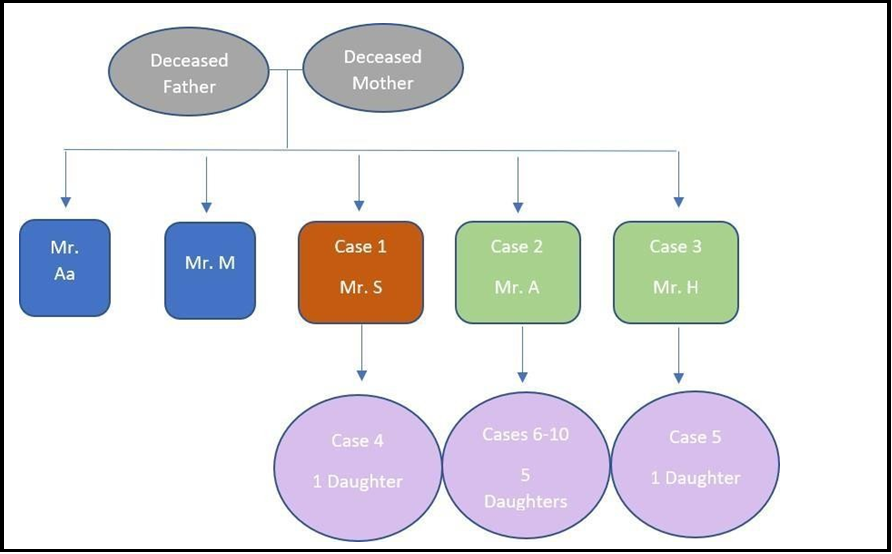
Figure 3: Family Pedigree
Case 2
Mr. A is a 46-year-old gentleman with hypertension and ESKD on regular hemodialysis. He is one of the affected brothers who tested positive for Fabry disease. No past medical history of cardiovascular disease or cerebrovascular events was documented.
Case 3
Mr. H is a 49-year-old gentleman with ESKD on regular hemodialysis and valvular heart disease post valve replacement and pacemaker insertion. He is the other affected brother who tested positive for Fabry disease. Apart from his cardiac and renal conditions, no other organs were affected.
Case 4
Ms. ES, the daughter of the index case, was screened for Fabry disease at the age of 15 and tested positive. She did not show any evidence of organ involvement at that time. At 17, she was referred for cardiac evaluation by echocardiogram as part of screening. The echocardiogram showed a mildly dilated left atrium with trace mitral valve regurgitation. Otherwise, the rest of the study was unremarkable, with an average ejection fraction of > 55 %. She was also referred to the dermatology service to assess the multiple skin lesions on her right arm, which had been there since birth. They thought these lesions were melanocytic nevus vs. nevus spills with no indication of a skin biopsy. Meanwhile, Ms. ES is still following up with our nephrology service for microscopic hematuria and stable kidney function, and a kidney biopsy is planned for her. On Ultrasound, both her kidneys were normal in size with preserved parenchymal echogenicity.
Case 5
Ms. SH is the daughter of Mr.H, who is 26 years old and under follow- up with our nephrology center for managing her microscopic hematuria and proteinuria, and she was referred for a kidney biopsy. Her echocardiogram was unremarkable. Despite no other organ being affected, she is on regular follow-ups with the dermatologist, ophthalmologist, and neurologist.
Cases from (6 – 10)
Mr. A, the other brother, has five daughters, all of whom tested positive during screening. Four of them are under the care of adult nephrology for managing microscopic hematuria and albuminuria with stable renal function tests. The fifth daughter is still under follow-up with pediatric nephrology. Their cardiac evaluations are unremarkable, with typical echocardiogram results. No other disease manifestations were detected during follow-up. (Table 1) illustrates the patients’ characteristics for the ten cases.
Due to the unavailability of enzyme replacement therapy in our center, all ten cases were referred to another local hospital to initiate the appropriate treatment.
Table 1: Clinical characteristics of the studied population
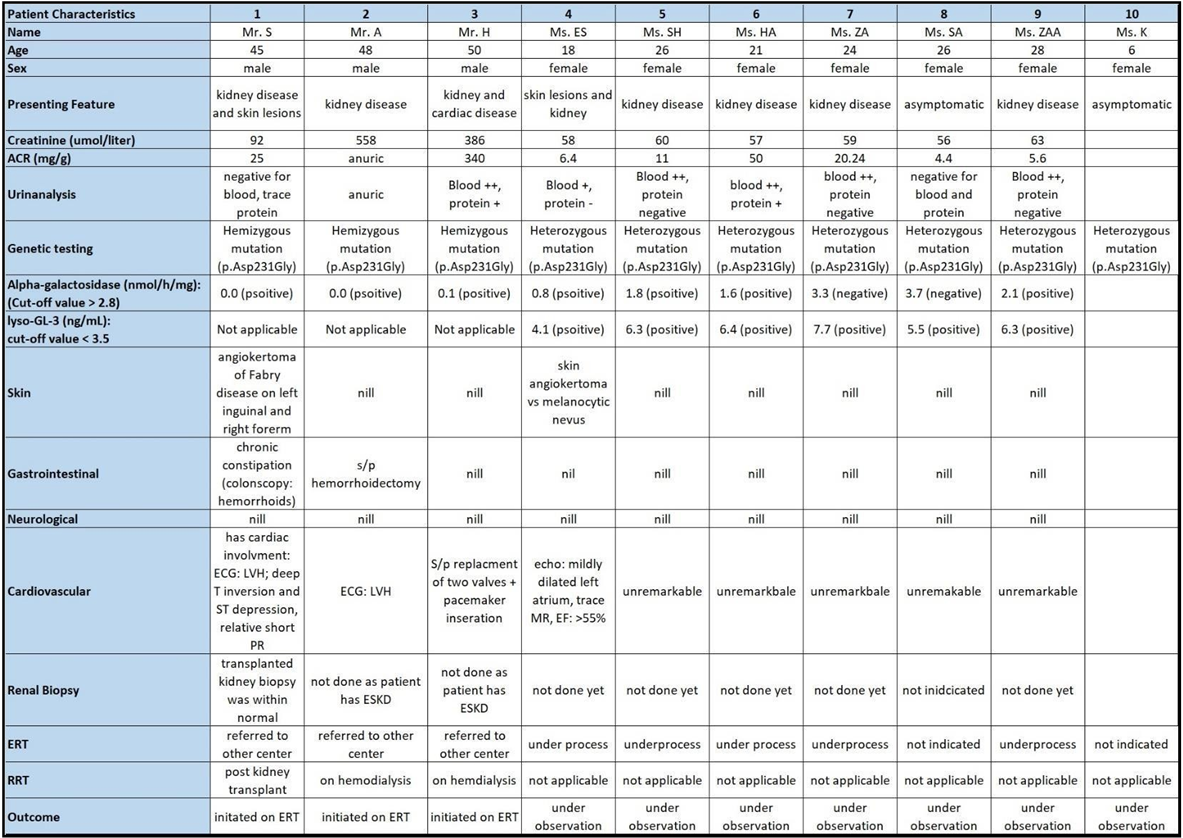
ACR: Albumin-Creatinine Ratio, ERT: Enzyme Replacement Therapy, RRT: Renal Replacement Therapy
Discussion
Epidemiology
Fabry disease is an X-linked rare lysosomal storage disorder resulting from the deficiency of α-galactosidase A (α-Gal A) and affects multiple systems. The prevalence of Fabry disease has been estimated between 1:40,000 and 1:60,000, while in other registries, it reached approximately 1:117,000 between individuals. However, the majority is still underestimated and varies among different populations [9]. In the Russian nationwide screening program, for example, the prevalence of Fabry disease in 5,572 hemodialysis patients was 0.36 %, with a 10-fold increase in males than females (0.53 and 0.05 %, respectively) [10]. FD is thought to be underdiagnosed in Arab countries due to the limited publications in this region and the fact that few Arab patients with FD are included in the Fabry registry [2,6].
Pathogenesis
Fabry disease is caused by a deficiency or absence of α-galactosidase A (α-gal A) enzyme activity due to mutations in the GLA gene resulting in intracellular accumulation of globotriaosylceramide (Gb3) and its derivative leading to multiorgan damage [9]. Various cells are affected, including capillary endothelial cells, renal (podocytes, tubular cells, glomerular endothelial, mesangial, and interstitial cells), cardiac (cardiomyocytes and fibroblasts), and nerve cells [11].
Clinical Presentation
The primary disease process begins in infancy or earlier during fetal development. However, contrary to many other lysosomal storage diseases, most patients remain clinically asymptomatic during the very first years of life [8].
The Fabry disease was initially described in males with a severe clinical phenotype that develops early in childhood or adolescence and progresses to multiorgan failure, now known as “classic” Fabry disease. However, a larger group of patients produce later-onset phenotypes with a milder presentation called the “variant” phenotype [2,9]. Clinical presentation in females ranges from asymptomatic to severe phenotype, explained by the X chromosome inactivation (Lyonization) profile, but usually develops ten years later than in male patients [12]. Although many female carriers will be relatively asymptomatic, recent evidence has emerged that females with Fabry disease can develop clinical symptoms and have a reduced life expectancy compared with healthy subjects [13].
In classic Fabry disease, early manifestations occur during childhood and affect the peripheral nervous system, characterized by chronic neuropathic pain and episodic severe pain crises. Progressive hearing loss has been reported in 16-54 % of affected patients, while varying degrees of tinnitus presented as an early symptom [8,14,15]. Other symptoms such as sweating abnormalities (hypohidrosis), skin manifestation (angiokeratomas), gastrointestinal (GI) disturbances (bloating, diarrhea, abdominal pain), corneal (cornea verticillate) and lenticular opacities have also been reported at a young age [2,8,9,16]. Renal impairment, manifested by microalbuminuria and proteinuria, usually occurs in the 2nd to 3rd decade of life and progresses to renal failure [17]. Cardiac abnormalities involved a wide range of manifestations, including left ventricular hypertrophy (LVH), valvular dysfunction, electrocardiogram abnormalities, arrhythmias, ischemia, and cardiac fibrosis and were reported in approximately 40- 60 % of patients with Fabry disease [8,18]. Cerebrovascular involvement includes symptoms such as headache, dizziness, transient ischemic attacks, and ischemic strokes [19,20].
Diagnosis
Recognizing early signs and symptoms of the disease is essential to initiate the treatment in Enzyme Replacement Therapy (ERT) promptly and start screening the family members. However, early diagnosis of Fabry disease could be challenging as the clinical manifestations may resemble other disorders. Moreover, significant cardiac or renal dysfunction usually occurs later in adulthood; therefore, confirmation of the disease is traditionally delayed [8].
α-Gal A activity testing in plasma or leukocytes is diagnostic for male patients; however, in female patients, demonstration of the plasma enzyme activity is often found within the normal range. Therefore, genetic analysis is warranted to establish the disease. Screening individuals with a family history of Fabry disease is a helpful tool to identify patients before developing symptoms [2,8]. Furthermore, screening of the following high-risk groups may be beneficial to detect the disease early and start the management on time: patients with unexplained hypertrophic cardiomyopathy HCM or with unexplained LVH, patients with ESKD, post-kidney transplantation, or patients with unexplained proteinuria or microalbuminuria, individuals (aged 15–55 years) with unexplained stroke [9].
Sodré et al. reported, in a cross-sectional study, a prevalence of Fabry disease of 9.47 % in relatives of 71 patients with chronic kidney disease (CKD) [21]. The European Renal Best Practice (ERBP) has provided, based on a combination of systematic literature reviews, a consensus meeting with an international panel of experts, and peer review, guidance for nephrologists in diagnosing and treating Fabry nephropathy. Accordingly, it was suggested to screen patients for Fabry disease when there is an unexplained chronic kidney disease in males younger than 50 years and females of any age by measuring α- galactosidase A activity in plasma, white blood cells, or dried blood spots in men and mutation analysis in women [22].
In the pediatric population, the diagnosis of FD is infrequent due to the nonspecific nature of clinical presentation at a young age [23]. An interval of 13.7 years was documented between the onset of symptoms and diagnosis in men and 16.3 years in women [24], and the frequency of misdiagnosis reached approximately 25 % [25].
Treatment
Management of patients with Fabry disease should focus on supportive care to prevent further progression to irreversible tissue damage. ERT should be provided with proper follow-up and assessment of treatment response by a physician with experience in Fabry disease management [2]. ERT is available in the form of agalsidase alfa (Replagal)®, Shire HGT, Inc., Cambridge, MA, USA) and agalsidase beta (Fabrazyme®, Sanofi Genzyme, Cambridge, MA, USA). Agalsidase alfa is given at 0.2 mg/kg body weight every other week by intravenous (IV) infusion and is approved in many countries worldwide but not by the US Food and Drug Administration [26]. Agalsidase beta is administered at 1.0 mg/kg body weight once every 2 weeks as an IV infusion and is approved in Europe, the USA, and many other countries [27].
Enzyme replacement therapy has challenges. It is expensive (~$200,000 per patient annually) and time-consuming (i.e., hours required for infusion). Furthermore, ERT may cause infusion reactions and is ineffective in all patients, such as those with End Stage Organ Disease or with antibodies to the recombinant enzyme [28].
Clinical evidence supported the efficacy of ERT in female patients in improving cardiac parameters and quality of life and reducing the enzyme level. However, there were insufficient data to conclude the effects of ERT on the nervous system, gastrointestinal manifestations, and pain in female patients with Fabry disease [8,13]. In children, enzyme replacement therapy is considered to limit or prevent irreversible tissue damage and minimize disease symptoms. The two forms of enzyme therapy, agalsidase alfa (ALFA) and agalsidase beta (BETA), were approved from 7 and 8 years of age, respectively [23].
Outcomes of Kidney Transplantation in Fabry Disease
According to the European Renal Best Practice, ERT is not suggested for renal indications after renal transplantation but can be continued for non-renal presentations [22]. Current evidence on the outcomes of kidney transplantation in Fabry disease is limited, and thus challenges in clinical practice are increased. However, a systematic review and meta-analysis by Suarez et al. showed no significant differences in the risks of all-cause graft failure or mortality among recipients with vs. without Fabry disease despite the recurrence of the disease in 11.1 % of the cases during a median follow-up from 3-11.5 years [1]
Conclusion
Fabry disease is a rare progressive lysosomal storage disorder characterized by a deficiency of α-galactosidase A (α-gal A). It affects multiple systems, including the kidneys leading to End Stage Kidney Disease. The prevalence of FD is considered underdiagnosed, especially in Arab countries. To our knowledge, this is the third publication from Saudi Arabia. Herewith, we have presented a case series from one family from the Eastern Province of Saudi Arabia in whom they tested positive for Fabry disease during screening by enzyme assay. The index case has been investigated post-kidney transplantation, and accordingly, the family members were screened and evaluated.
Screening for Fabry disease is essential, especially in neonates, dialysis patients, and transplant populations, to identify the size of the problem in the Gulf and to implement early interventions for this disorder. It is also essential to acquire a national registry to understand better the disease characteristics and response to treatment in the Gulf region.
Acknowledgment
The authors are pleased to express their great gratitude and appreciation to Dr. Miral Mashhour from the pathology department at King Fahad Specialist Hospital - Dammam for her valuable contribution with the histopathological slides.
Ethical Approval
Ethical approval was obtained from King Fahad Specialist Hospital - Dammam Review Board.
Conflict of Interest
The authors have declared no potential conflicts of interest with respect to the case-series, authorship, and/or publication of this article.
References
- Suarez MLG, Thongprayoon C, Hansrivijit P, Medaura J, Vaitla P, et al. (2020) Outcomes of kidney transplantation in Fabry disease: a meta-analysis. Diseases. 9(1): 2.
- Ortiz A, Germain DP, Desnick RJ, Politei J, Mauer M, et al. (2018) Fabry disease revisited: management and treatment recommendations for adult patients. Molecular genetics and metabolism. 123(4): 416-27.
- Paim-Marques L, de Oliveira RJ, Appenzeller S (2022) Multidisciplinary Management of Fabry Disease: Current Perspectives. Journal of Multidisciplinary Healthcare. 15: 485- 495.
- Al-Jasmi FA, Tawfig N, Berniah A, Ali BR, Taleb M, et al. (2013) Prevalence and novel mutations of lysosomal storage disorders in United Arab Emirates. JIMD Reports. 10: 1-9.
- Al-Sannaa NA, Al-Abdulwahed HY, Al-Ghamdi MS (2017) Lysosomal storage disorders (LSDs): the prevalence in the eastern Province of Saudi Arabia. Int J Neurol Dis. 1(2): 038-043.
- Alhemyadi SA, Elawad M, Fourtounas K, Abdrabbou Z, Alaraki B, et al. (2020) Screening for Fabry disease among 619 hemodialysis patients in Saudi Arabia. Saudi Medical Journal. 41(8): 813-818.
- Nampoothiri S, Yesodharan D, Bhattacherjee A, Ahamed H, Puri RD, et al. (2020) Fabry disease in India: A multicenter study of the clinical and mutation spectrum in 54 patients. JIMD reports. 56(1): 82-94.
- Germain DP (2010) Fabry disease. Orphanet journal of rare diseases. 5: 30.
- Vardarli I, Rischpler C, Herrmann K, Weidemann F (2020) Diagnosis and screening of patients with Fabry disease. Therapeutics and clinical risk management. 16: 551-558.
- Moiseev S, Fomin V, Savostyanov K, Pushkov A, Moiseev A, et al. (2019) The prevalence and clinical features of Fabry disease in hemodialysis patients: Russian Nationwide Fabry Dialysis screening program. Nephron. 141(4): 249-55.
- Desnick RJ, Ioannou YA, Eng CM (2001) Alpha-galactosidase A deficiency: Fabry disease. In The metabolic and molecular bases of inherited disease. 3733-3774.
- Torra R (2008) Renal manifestations in Fabry disease and therapeutic options: New strategies to prevent cardiovascular risk in chronic kidney disease. Kidney International. 74: S29-32.
- Germain DP, Arad M, Burlina A, Elliott PM, Falissard B, et al. (2019) The effect of enzyme replacement therapy on clinical outcomes in female patients with Fabry disease–A systematic literature review by a European panel of experts. Molecular genetics and metabolism. 126(3): 224-35.
- Germain DP, Avan P, Chassaing A, Bonfils P (2002) Patients affected with Fabry disease have an increased incidence of progressive hearing loss and sudden deafness: an investigation of twenty-two hemizygous male patients. BMC medical genetics. 3: 10.
- Hegemann S, Hajioff D, Conti G, Beck M, Sunder-Plassmann G, et al. (2006) Hearing loss in Fabry disease: data from the Fabry Outcome Survey. European journal of clinical investigation. 36(9): 654-62.
- Zarate YA, Hopkin RJ (2008) Fabry's disease. The Lancet. 372(9647): 1427-35.
- Ortiz A, Oliveira JP, Waldek S, Warnock DG, Cianciaruso B, et al. (2008) Nephropathy in males and females with Fabry disease: cross-sectional description of patients before treatment with enzyme replacement therapy. Nephrology Dialysis Transplantation. 23(5): 1600-7.
- Senechal M, Germain DP (2003) Fabry disease: a functional and anatomical study of cardiac manifestations in 20 hemizygous male patients. Clinical genetics. 63(1): 46-52.
- Mitsias P, Levine SR (1996) Cerebrovascular complications of Fabry's disease. Annals of Neurology. 40(1): 8-17.
- Wozniak MA, Kittner SJ, Tuhrim S, Cole JW, Stern B, Dobbins M, et al. (2010) Frequency of unrecognized Fabry disease among young European-American and African-American men with first ischemic stroke. Stroke. 41(1): 78-81.
- Sodré LSS, Huaira RMNH, Colugnati FAB, Carminatti M, Braga LSS, et al. (2020) Screening of family members of chronic kidney disease patients with Fabry disease mutations: a very important and underrated task. Brazilian Journal of Nephrology. 43(1): 28- 33.
- Terryn W, Cochat P, Froissart R, Ortiz A, Pirson Y, et al. (2013) Fabry nephropathy: indications for screening and guidance for diagnosis and treatment by the European Renal Best Practice. Nephrology dialysis transplantation. 28(3): 505-17.
- Vaisbich MH, Andrade LG, Silva CA, Barreto FD (2022) Recommendations for the diagnosis and management of Fabry disease in pediatric patients: a document from the Rare Diseases Committee of the Brazilian Society of Nephrology (Comdora- SBN). Brazilian Journal of Nephrology. 44(2): 268-280.
- Mehta A, Ricci R, Widmer U, Dehout F, Garcia de Lorenzo A, et al. (2004) Fabry disease defined: baseline clinical manifestations of 366 patients in the Fabry Outcome Survey. European journal of clinical investigation. 34(3): 236-42.
- Germain DP, Fouilhoux A, Decramer S, Tardieu M, Pillet P, et al. (2019) Consensus recommendations for diagnosis, management and treatment of Fabry disease in paediatric patients. Clinical genetics. 96(2): 107-117.
- Replagal® Summary of Product Characteristics, Available at: http://www.ema.europa.eu.
- Fabrazyme® Summary of Product Characteristics, Available at: http://www.ema.europa.eu.
- Miller JJ, Kanack AJ, Dahms NM (2020) Progress in the understanding and treatment of Fabry disease. Biochimica et Biophysica Acta (BBA)-General Subjects. 1864(1): 129437.