


Inusha Panigrahi*, Shalini Dhiman, Parminder Kaur, Priyanka Srivastava
Genetic Metabolic Unit, Department of Pediatrics, Post Graduate Institute of Medical Education and Research, India
*Corresponding Author: Inusha Panigrahi, Genetic Metabolic Unit, Department of Pediatrics, Post Graduate Institute of Medical Education and Research, India.
Abstract
Rubinstein–Taybi syndrome (RSTS) is a genetic disorder characterized by intellectual disability, distinctive facial features, growth retardation, and limb anomalies. Pathogenic variants in CREBBP and EP300 are the primary molecular causes of RSTS. We presented clinical and genetic data of six children diagnosed with RSTS at our single tertiary care centre. All underwent detailed phenotypic assessment and genetic testing, including “multiplex ligation-dependent probe amplification (MLPA), and whole exome sequencing (WES)”. Six children, aged 1–13 years, presented to the Genetics Clinic with developmental delay, intellectual disability, and dysmorphic craniofacial features. One child had broad thumbs, and another had 2–3 toe syndactyly. Three patients harbored truncating single-nucleotide variants (SNVs) in CREBBP (exons 10, 14, and 22), and one had a heterozygous 16p13.3 deletion detected by MLPA. RSTS is an important and relatively frequent cause of developmental delay and intellectual disability in children with characteristic dysmorphism. Early recognition of clinical clues, followed by appropriate genetic testing, enables timely genetic counseling, diagnosis, and management.
Keywords: Rubinstein–Taybi syndrome, CREBBP, intellectual disability, single-nucleotide variant, copy number variant, dysmorphism
Introduction
Rubinstein–Taybi syndrome (RSTS) is a rare, clinically recognizable autosomal dominant disorder first linked in 1992 to genetic alterations involving chromosome 16, including chromosomal breakpoints, point mutations, and microdeletions. The estimated birth prevalence ranges from 1 in 100,000 to 1 in 125,000 live births worldwide [1–3]. Despite its rarity, RSTS remains an important cause of syndromic intellectual disability encountered in pediatric and clinical genetics practice.
RSTS is divided into two genetically defined subtypes: type 1 (RSTS1; OMIM #180849), caused by heterozygous variation in the CREBBP gene, and type 2 (RSTS2; OMIM #613684), caused by heterozygous variantion in the EP300 gene. Collectively, CREBBP variants account for approximately 50–60% of clinically diagnosed cases, whereas EP300 variants contribute to 5–8% [4–7]. In roughly 30% of patients fulfilling the clinical criteria for RSTS, the underlying molecular variation remains unidentified, suggesting the possibility of additional genes or non-coding regulatory variants yet to be discovered.
Clinically, RSTS is characterized by distinctive features: microcephaly, moderate to severe intellectual disability, postnatal growth retardation, and characteristic craniofacial dysmorphism, often accompanied by a “grimacing” smile. Limb anomalies, most notably broad thumbs and broad halluces are considered hallmark findings [1–3]. Associated anomalies are frequent and may involve multiple systems, including ocular defects (e.g., cataracts, coloboma), congenital heart disease, renal malformations, cryptorchidism in males, and skeletal anomalies. The clinical phenotype is heterogeneous, and the full spectrum of features may not be apparent in infancy, often delaying diagnosis.
The most of cases arise sporadically due to de novo pathogenic variants. These may include small deletions or insertions leading to premature termination codons, splicing defects, or missense changes affecting protein function, as well as large deletions, pericentric inversions, and balanced or unbalanced translocations involving CREBBP or EP300 [8–11]. To date, more than 500 pathogenic CREBBP variants and over 100 EP300 variants have been documented, distributed across 31 exons. Variant spectrum analysis reveals a predominance of missense changes (76–80%), followed by frameshift (6–10%), nonsense (6–8%), and splice-site variants (3–5%) [12–13].
RSTS belongs to the group of chromatinopathies, Mendelian disorders caused by defects in genes encoding chromatin transcriptional regulators resulting in epigenetic dysregulation during development. This pathophysiological basis explains the phenotypic overlaps with other syndromes such as Cornelia de Lange syndrome, Kabuki syndrome, Wiedemann–Steiner syndrome, Genitopatellar syndrome, Floating Harbor syndrome. Careful clinical evaluation combined with targeted molecular testing is essential to distinguish these entities, given their overlapping features but differing prognoses and recurrence risks [14-18].
In this report, we present four Asian Indian children with CREBBP- related RSTS, including three harboring truncating single-nucleotide variants and one with a pathogenic copy number variant at 16p13.3. We detail their clinical phenotypes, neuroimaging findings, and genetic results, and discuss the diagnostic approach, genotype– phenotype correlations, and implications for genetic counseling.
Cases Presentation
Case 1: A 1.5-year-old male child was referred to the Genetics Clinic for evaluation of global developmental delay, with recent onset of fever, episodes of vacant staring, and vomiting. He was the first child of healthy, non-consanguineous parents. The birth weight of the child was 2.5 kg following an uneventful antenatal course and delivery. There was no family history of neurodevelopmental disorders, congenital anomalies, or genetic syndromes.
At presentation, anthropometric measurements showed a weight of – 2.4 Z score and occipitofrontal circumference (OFC) of –3.39 Z score, indicating significant microcephaly and growth restriction. Physical examination revealed multiple dysmorphic features, including light, sparse scalp hair; grey, deep-set eyes; an elongated skull; prominent ears; a high-arched palate; and retrognathia (Figure 1a). Neurological examination confirmed global developmental delay.
Magnetic resonance imaging (MRI) of the brain demonstrated multiple micro-hemorrhages in the left basal ganglia and right cerebellar hemisphere, as well as loss of flow void in the straight sinus and right transverse sinus, suggestive of venous sinus involvement. Ophthalmological evaluation, including fundus examination, was normal. Hearing assessment and a complete skeletal survey showed no abnormalities. Cardiovascular examination and echocardiography excluded congenital heart defects.
Given the combination of microcephaly, growth retardation, and dysmorphic craniofacial features, a syndromic etiology was considered, with a provisional differential diagnosis of a chromosomal deletion–duplication syndrome. Informed written consent was obtained from the parents for clinical photography and genetic testing.
Molecular analysis for copy number changes was performed using the P245 MLPA kit (MRC-Holland), which screens for known microdeletions and microduplications in multiple syndromic loci, including CREBBP. The assay revealed a heterozygous deletion involving exon 1 of the CREBBP gene at chromosome 16p13.3, confirming the diagnosis of RSTS1. This finding was consistent with the clinical phenotype and provided a definitive molecular diagnosis, enabling appropriate genetic counseling for the family.
Case 2: A 1-year-old girl presented with dysmorphic craniofacial features, including a broad forehead and wide nasal bridge, along with multiple limb anomalies polydactyly of the left foot, syndactyly, sclerosteosis, and hyperphalangy. Based on her clinical presentation, Menke–Hennekam syndrome type 1 was suspected. Molecular analysis by WES revealed a heterozygous, pathogenic splice-site variant in the CREBBP gene, located on chromosome 16: NM_004380.3 (CREBBP): c.4890+1G>A. This variant affects the canonical splice donor site, likely resulting in aberrant mRNA splicing and loss of normal CREBBP protein function.
Case 3: A 2-year-old male was evaluated for developmental concerns. The child exhibited intellectual disability and global developmental delay with significant delays in achieving gross motor and speech milestones. On physical examination, craniofacial dysmorphism was noted, including dysplastic ears and downslanting palpebral fissures. The extremity examination revealed broad thumbs, a classical phenotypic feature of RSTS. Genetic testing was pursued after an MLPA result. WES identified a heterozygous nonsense variant in the CREBBP: c.1977C>A, resulting in the premature stop codon p. Tyr659*. This variant is located in exon 10 of the CREBBP gene and is predicted to result in a truncated protein lacking critical functional domains, thereby disrupting the transcriptional coactivator activity of CREBBP.
Case 4: A 1-year-old male was evaluated for global intellectual disability and developmental delay. He was born to non- consanguineous parents. Physical examination revealed brachycephaly, hypospadias, and bilateral undescended testes. Neuroimaging by MRI demonstrated complete agenesis of the corpus callosum. Genetic testing using targeted sequencing of the CREBBP gene identified a heterozygous nonsense variant, CREBBP: c.3906G>A (p. Trp1302*), located in exon 22. The change was predicted to introduce a premature stop codon, likely resulting in a truncated protein. The clinical and molecular findings supported a diagnosis of RSTS1.
Case 5: A 2.5-year-old male presented with intellectual disability and microcephaly. He was the first child of healthy, non-consanguineous parents. Clinical examination revealed syndactyly of the 2nd and 3rd toes, hypoplastic toenails, and dysmorphic facial features. Additional systemic evaluation identified an atrial septal defect on echocardiography and unilateral hydronephrosis on abdominal ultrasonography. The facial profiles of cases 3 and 4 are shown in Figure 1(b, d).
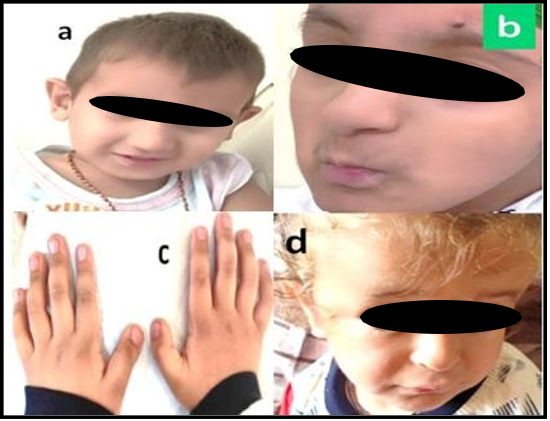
Figure 1: Facial profile of children with Rubinstein-Taybi syndrome showing prominent nose, overhanging columella, large ears, and thin upper lip; note the grimace in 1a. The brachydactyly and broad thumbs in case 3 is shown in 1c.
Genetic analysis identified a heterozygous nonsense variant in the CREBBP: c.2767C>T (p. Gln923*), located in exon 14. This change is predicted to result in a truncated protein and is consistent with a molecular diagnosis of RSTS1.
Case 6: A 13-year-old male presented with a history of intellectual disability and increased anxiety, along with difficulties in social adaptation and learning. Physical examination revealed short stature (height below the 3rd percentile for age) and coarse facial features. Distinct craniofacial findings included a prominent nose and retrognathia.
Following a negative MLPA result, WES was performed. Genomic interpretation identified a heterozygous, missense variant in the EP300 gene: c.6842A>G, resulting in an amino acid substitution p. Gln2281Arg. This variant lies within a functionally important region of the EP300 protein, which, like CREBBP, acts as a transcriptional coactivator with histone acetyltransferase activity.
The phenotypic and genotypic details of children are presented in Table 1.
Table 1: Clinical characteristics of patients with Rubinstein-Taybi syndrome and single-nucleotide variants in the CREBBP gene in the present report.
|
S. No |
Age/Sex |
Clinical features |
Variant description |
Variant type |
|
C1 |
1.5years/ M |
Global developmental delay, dysmorphic |
CREBBP exon 1 deletion Pathogenic |
Pathogenic (Detected by |
|
C2 |
1 year/F |
Wide nasal bridge, polydactyly of the left foot, syndactyly, sclerosteosis, |
CREBBP: c.4890+1G>A Exon 29 Splice |
Pathogenic rs1596793242 |
|
C3 |
2year/M |
Global developmental delay, intellectual |
CREBBP: c.1977C>A (p. Tyr659*) Exon10 |
Pathogenic rs2053190428 |
|
C4 |
1year/M |
Global developmental delay, brachycephaly, |
CREBBP:c.3906G>A(p.Trp1302*) Exon22 |
Likely Pathogenic rs2151355361 |
|
C5 |
2.5 years/M |
Intellectual disability, microcephaly, |
CREBBP: c.2767C>T(p.Gln923*) Exon 14 |
Likely Pathogenic rs2141200227 |
|
C6 |
13 years/M |
Intellectual disability, increased anxiety, |
EP300:c.6842 A>G (p.Gln2281Arg) |
Variant of unknown |
WES identified splice site and truncating variants in the CREBBP gene in the four children, while a VUS in the EP300 gene was detected in one child (Figure 2). The CREBBP variants were absent in the Genome Aggregation Database (gnomAD) and were predicted to be deleterious or of high functional impact by multiple in silico tools, including SIFT, LRT-Pred, PolyPhen-2, REVEL, and MetaSVM.Prenatal diagnosis was subsequently performed in one affected family using targeted variant analysis, and the fetus was found to be unaffected.
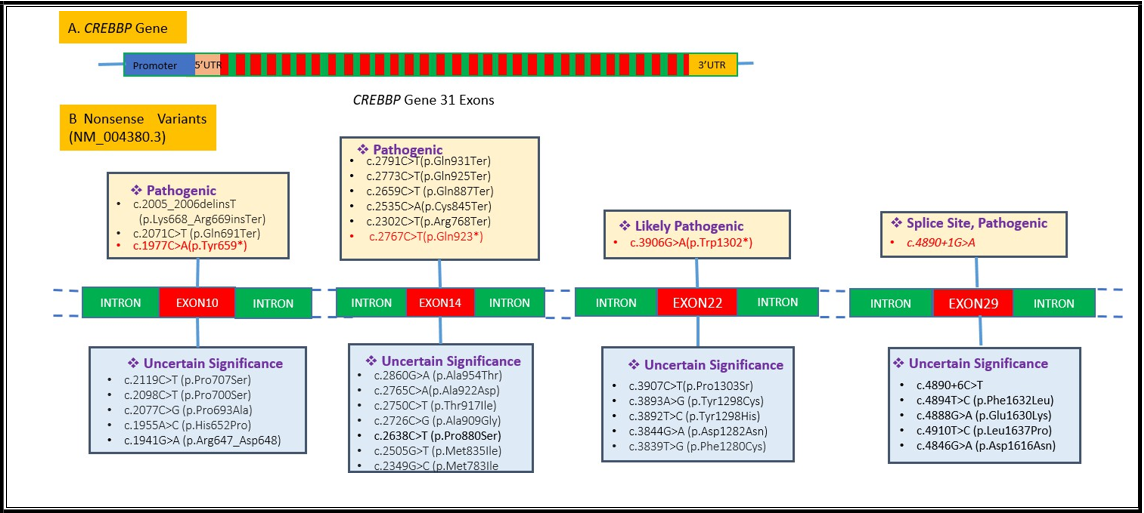 Figure 2: Reported variants in the CREBBP gene in exons 10,14, 22, and 29 are illustrated here; the single-nucleotide variants in the present study are shown below in red color.
Figure 2: Reported variants in the CREBBP gene in exons 10,14, 22, and 29 are illustrated here; the single-nucleotide variants in the present study are shown below in red color.
The distribution of pathogenic variant types in the CREBBP and EP300 genes among patients with RSTS is illustrated in Figure 3. In the CREBBP cohort, missense variants were most prevalent (76%), followed by frameshift (12%), nonsense (8%), and splice-site variants (4%). In the EP300 cohort, missense variants predominated (80%), with frameshift (11%), splice-site (6%), and nonsense variants (3%) observed at lower frequencies. These findings highlight the predominance of missense changes in both genes, though CREBBP exhibits a slightly higher proportion of truncating variants compared to EP300.
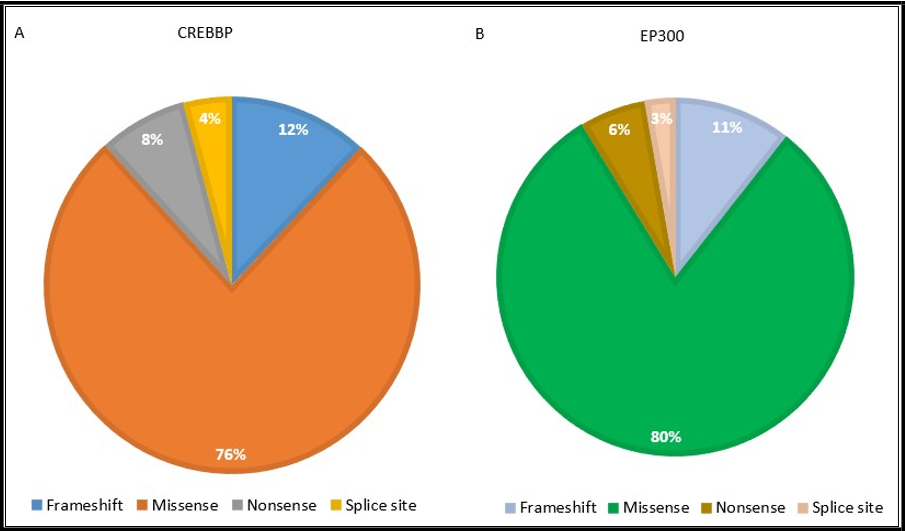
Figure 3: Distribution of the variants (Frameshift, missense, nonsense, splice site) in CREBBP and EP300.
In silico analysis of the RSTS Genes:
CREBBP binding proteins acetylate histones and act as a specific transcriptional activation marker. CREBBP is acetylates the non- histone proteins like MYB, NOCOA1, NCOA3, TP53, HIFIA, RELA, KAT2B, STAT1 and FOXO1 (Figure 4). CREB1 is aphosphorylation-dependent transcription factor that is activated in response to various extracellular signals, including cAMP, calcium, and growth factors. Upon phosphorylation at Ser133, CREB1 binds to CRE sequences within target gene promoters and recruits coactivators such as EP300. This interaction facilitates chromatin relaxation through histone acetylation, thereby promoting transcription of genes involved in cell growth, neuronal differentiation, memory formation, and other signaling pathways [4-8].
The EP300 protein functions as a transcriptional coactivator with intrinsic histone acetyltransferase (HAT) activity. It plays a pivotal role in regulating transcription via chromatin remodeling.
Specifically, EP300 binds to phosphorylated CREB1 (cyclic AMP- responsive element-binding protein 1) and enhances its transcriptional activation of genes containing the CRE (cAMP- responsive element) sequence [6-10]. The interactome of CREBBP and EP300 protein is created by String database. Pathogenic and uncertain significance variants found on the CREBBP and Ep300 variant significance was classified by ACMG Criteria and deleterious potential was evaluated by the SIFT, Mutation taster, PolyPhen 2 databases. Variants were described according to HGVS nomenclature guidelines [10].
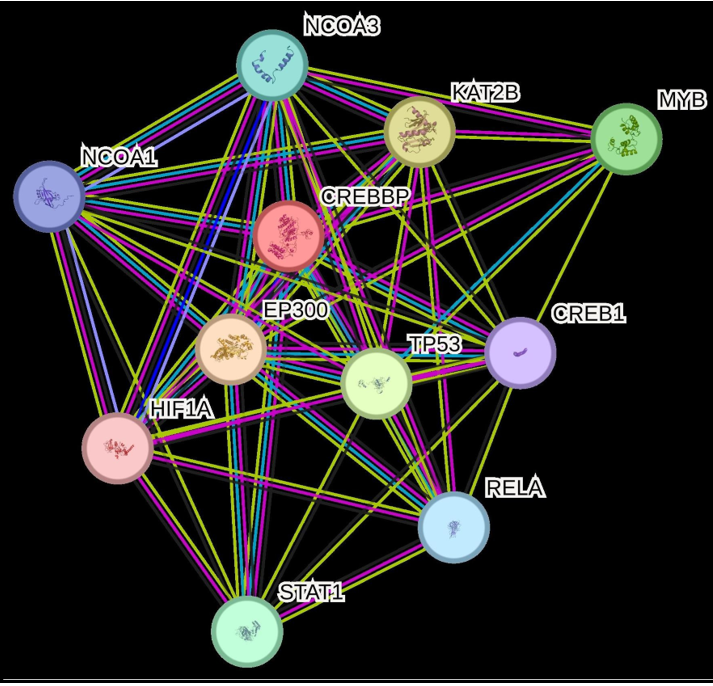
Figure 4: CREBBP interactome with acetylates histones and non-histone proteins.
Discussion
RSTS is a genetically heterogeneous disorder primarily caused by pathogenic variants in CREBBP and, less frequently, EP300, both of which encode transcriptional coactivators with intrinsic histone acetyltransferase (HAT) activity. These proteins play a major role in chromatin remodeling and the regulation of gene transcription via interaction with multiple transcription factors, including CREB, p53, and nuclear hormone receptors. Variants in CREBBP account for approximately 50–60% of RSTS cases, with more than 500 pathogenic variants reported to date across population studies and mutation databases. EP300 variants contribute to 8–15% of cases, with over 100 pathogenic variants documented [1-6].
The mutational spectrum in CREBBP is dominated by missense variants (76–80%), which can disrupt protein stability, protein–protein interactions, or HAT activity, leading to transcriptional dysregulation of developmental genes. Frameshift (6–10%), nonsense (6–8%), and canonical splice site variants (3–5%) typically result in haploinsufficiency via premature truncation or aberrant splicing, thereby reducing functional protein dosage. EP300-associated RSTS tends to display milder phenotypic manifestations, potentially due to partial functional redundancy between EP300 and CREBBP, though both genes are essential for normal embryogenesis and organogenesis.
Clinically, RSTS presents with well-recognized features as craniofacial, skeletal, and growth abnormalities. The craniofacial phenotype characterized by down-slanting palpebral fissures, microcephaly, abnormal auricular morphology, micrognathia, and a prominent philtrum largely reflects aberrant development of neural crest cell–derived craniofacial structures, a process regulated in part by the CREBBP and EP300 coactivators through modulation of chromatin remodeling and transcriptional programs during embryogenesis. Neural crest–related defects may also underlie the distinctive auricular positioning and mandibular hypoplasia.
In our cohort, additional facial traits such as arched eyebrows, long eyelashes, anteverted large ears, and a thin upper lip were noted. These features align with phenotypic spectra reported in previous literature, further underscoring the variability within the RSTS craniofacial presentation. The broad thumbs and halluces observed in most patients likely result from disrupted signaling pathways in limb bud morphogenesis, possibly involving BMP and WNT pathways downstream of CREBBP/EP300 transcriptional regulation. The postnatal growth retardation common in RSTS may reflect systemic effects of these transcriptional dysregulations on growth plate chondrocyte proliferation and differentiation.
Skeletal anomalies such as short, broad thumbs and toes are thought to arise from dysregulated expression of HOX and other limb- patterning genes downstream of CREBBP/EP300-mediated acetylation pathways. Cardiac defects, including atrial and ventricular septal defects and, less commonly, pulmonary stenosis, occur in approximately one-third of patients, likely reflecting disrupted regulation of cardiac morphogenesis genes (e.g., NKX2-5, TBX5). The coexistence of craniofacial, skeletal, and cardiac features underscores the pleiotropic developmental role of CREBBP and EP300, with pathogenic variants perturbing transcriptional programs across multiple germ layers [4-12].
However, no cardiac and skeletal anomalies were observed in the first patient in the present report. Renal abnormalities can also be observed, and hypospadias is seen in some patients. One child in the present report had atrial septal defect and hydronephrosis. The secondary risk factors associated are seizures and additional symptoms, such as behavioral problems and autistic features including other medical complications, have also been reported in previous literature. The patients of this syndrome are prone to develop certain tumors, typically found in children such as meningioma, leukemia, neuroblastoma, medulloblastoma, and rhabdomyosarcoma, although meningioma can also occur later in adulthood [14-18].
Two genes have been implicated in this syndrome: CREBBP and EP300 located on chromosome 16p13.3, and chromosome 22q13.2. Both genes have 31 exons, a high sequence similarity of about 70%, and function as transcriptional co-activators. They have effective histone acetyltransferase (HAT) activity and act by targeting H3 and H4 histones [8,9,11]. The histone acetylation unfolds the structure of the chromatin, which is essential for the expression of genes. CREBPP and EP300 acetylate a range of proteins, including p53. Previous studies show that reduced HAT activity is sufficient to cause the syndrome [12-14]. Though mostly sporadic, in a few cases, one of the parents might be an asymptomatic carrier of a pathogenic SNV. Previous studies have reported that large gene deletions in CREBBP contribute to severe RSTS phenotypes than those caused by SNV [19]. Multiple molecular techniques have been widely used to find the copy number variation, such as MLPA and chromosomal microarray (CMA). Nowadays, long-read generation sequencing technologies can also identify the copy number variants, in addition to single-nucleotide variants. MLPA can identify up to 93-95% deletions and insertions. Thus, MLPA is a cost- effective test for the identification of common copy number variations and can be used as a first-tier test for the diagnosis of the syndrome. Probably CREBBP is a common gene implicated in the causation of RSTS phenotype in Indian children, as it was found in 5 patients, whereas the EP300 variant was detected in one older patient with a milder phenotype.
Conclusions
RSTS is classified as one of the prototype syndromes with multiple congenital abnormalities and intellectual disability in clinical genetics, caused by dysregulation of gene expression. RSTS should be considered in children with intellectual disability and/or behavioral problems, even if additional malformations are not present. If abnormality at a cytogenetic or molecular level has been identified, proper genetic counseling and antenatal diagnosis is possible in affected families.
Conflicts Of Interest Statement: The authors have no conflicts of interest to declare.
Author Contributions:
IP: Drafting and revising the manuscript critically for important intellectual content and follow up of patient.
SD: Drafting and revising the manuscript, variant interpretation PK: Management and follow-up of patients.
PS: MLPA testing, variant interpretation
All authors have read and approve of final version of the manuscript. All authors agree to be accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Statement of Ethics: The report is in accordance with the Declaration of Helsinki. Written informed consent was taken from the guardian of the patient for the publication of images.
Acknowledgements: We thank all junior residents and others in the Genetic lab, who were involved in the evaluation of the patient.
References
- Awan N, Pearson E, Shelley L, Greenhill C, Tarver J, et al. (2022) The behavioral phenotype of Rubinstein–Taybi syndrome: A scoping review of the literature. American Journal of Medical Genetics Part A. 188(9): 2536-2554.
- Enomoto Y, Yokoi T, Tsurusaki Y, Murakami H, Tominaga M, et al. (2022) Divergent variant patterns among 19 patients with Rubinstein‐Taybi syndrome uncovered by comprehensive genetic analysis including whole genome sequencing. Clinical Genetics. 101(3): 335-345.
- Giani L, Michelini G, Ajmone PF, Scaini S, Selicorni A, et al. (2022) Age-related hallmarks of psychopathology in Cornelia de Lange and Rubinstein-Taybi syndromes. Research in Developmental Disabilities. 126(1): 104235.
- Kumar S, Suthar R, Panigrahi I, Marwaha RK (2012) Rubinstein- Taybi syndrome: Clinical profile of 11 patients and review of literature. Indian journal of human genetics. 18(2): 161.
- Levetan C, Van Gils J, Saba A, Rodríguez-Fonseca C, Fieggen, K, et al. (2022) Rubinstein-Taybi syndrome: presentation in the first month of life. The Journal of Pediatrics. 249(1): 106-110.
- López M, Seidel V, Santibáñez P, Cervera-Acedo C, Castro-de Castro P, et al. (2016) First case report of inherited Rubinstein- Taybi syndrome associated with a novel EP300 variant. BMC Medical Genetics. 17(1): 97.
- Negri G, Magini P, Milani D, Crippa M, Biamino E et al. (2019) Exploring by whole exome sequencing patients with initial diagnosis of Rubinstein–Taybi syndrome: the interconnections of epigenetic machinery disorders. Human Genetics. 138(3): 257- 269.
- Pérez-Grijalba V, García-Oguiza A, López M, Armstrong J, García-Miñaur S et al. (2019) New insights into genetic variant spectrum and genotype–phenotype correlations of Rubinstein‐ Taybi syndrome in 39 CREBBP‐positive patients. Molecular Genetics & Genomic Medicine. 7(11): e972.
- Tekendo-Ngongang C, Owosela B, Fleischer N, Addissie YA, Malonga B et al. (2020) Rubinstein–Taybi syndrome in diverse populations. American Journal of Medical Genetics Part A 182(12): 2939-2950.
- Richards S, Aziz N, Bale S, Bick D, Das S et al. (2015) Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genetics in medicine 17(5): 405-423.
- Vale AR, de Avó LRDS, Pilotto RF, Germano CMR, Melo DG (2022). Quality of life of Brazilian families who have children with Rubinstein–Taybi syndrome: An exploratory cross‐sectional study. American Journal of Medical Genetics Part A. 188(11): 3294-3305.
- Van Gils J, Magdinier F, Fergelot P, Lacombe D (2021) Rubinstein-Taybi syndrome: a model of epigenetic disorder. Genes. 12(7): 968.
- Waite J, Beck SR, Powis L, Oliver C (2023) The executive function account of repetitive behavior: evidence from Rubinstein-Taybi syndrome. American journal on intellectual and developmental disabilities. 128(1): 49-65.
- Wang Q, Wang C, Wei WB, Rong WN, Shi XY (2022) A novel CREBBP mutation and its phenotype in a case of Rubinstein– Taybi syndrome. BMC Medical Genomics. 15(1): 182.
- Fergelot P, Van Belzen M, Van Gils J, Afenjar A, Armour CM, et al. (2016) Phenotype and genotype in 52 patients with Rubinstein–Taybi syndrome caused by EP300 mutations. American Journal of Medical Genetics PartA 170(12): 3069-3082.
- Ellis K, Oliver C, Stefanidou C, Apperly I, Moss J (2020) An observational study of social interaction skills and behaviors in Cornelia de Lange, fragile X and Rubinstein-Taybi syndromes. Journal of autism and developmental disorders. 50(11): 4001-4010.
- Pérez-Grijalba V, García-Oguiza A, López M, Armstrong J, García-Miñaur S et al (2019) New insights into genetic variant spectrum and genotype–phenotype correlations of Rubinstein‐ Taybi syndrome in 39 CREBBP‐positive patients. Molecular Genetics & Genomic Medicine. 7(11): e972.
- Jin E, Le H, Jewell A, Couser NL (2024) Genotype-phenotype analysis of ocular findings in Rubinstein-Taybi syndrome–A case report and review of literature. Ophthalmic genetics. 45(1): 51- 58.