


Hallah Alanazi, Hayat Alrabieah, Lubna Khan*
King Abdul-Aziz medical city, Department of obstetrics and gynecology, reproductive medicine unit, Saudi Arabia, Riyadh.
*Corresponding Author: Lubna Khan, King Abdul-Aziz medical city, Department of obstetrics and gynecology, reproductive medicine unit, Saudi Arabia, Riyadh.
Abstract
Introduction
Congenital absence of Mullerian duct derivatives, known as Mayer Rokitansky Kuster Hauser syndrome is a rare condition, It is usually associated with urological and skeletal abnormalities. Its association with bilateral gonadal dysgenesis is extremely rare and appears to be independent of chromosomal abnormalities.
Case report
We are reporting a case of a 17-year-old girl who had presented with primary amenorrhea and the absence of secondary sexual characteristics. No other abnormalities were detected on examination. The endocrine evaluation revealed a picture of hypogonadotropic hypogonadism with normal thyroid function. Bone mineral density was less than expected for the patient's age. She has a normal female karyotype 46 XX. Pelvic ultrasound showed no uterus or ovaries. Pelvic MRI showed the absence of the uterus, cervix, vagina, and both ovaries which confirm the diagnosis of Mayer Rokitansky Kuster Hauser syndrome and bilateral ovarian dysgenesis. Brain MRI was normal. Urinary tract abnormalities were excluded as well by MRI.
Hormonal replacement therapy was started as conjugated estrogen to trigger her secondary sexual characteristics and osteoporosis prevention.
Conclusion
The association of Mayer Rokitansky Kuster Hauser syndrome and bilateral ovarian dysgenesis is a rare condition with unknown pathogenesis that is independent of chromosomal abnormalities. The management will be supportive with hormone replacement therapy to develop secondary sexual characteristics and to avoid premature bone loss. Unfortunately, there is nothing much that can be done to improve her fertility potential.
Keywords: MRKH, Gonadal Dysgenesis, fertility
Introduction
Gonadal dysgenesis in females is defined as absent or limited development of the ovaries. As a result of the hypogonadotropic hypogonadism hormonal status of such cases, patients will present with amenorrhea and lack of development of secondary sexual characteristics. It occurs in < 1 in 10,000 women with varied karyotyping which can be 46XX, 45XO, mosaicism, or deletion of a certain part of the X chromosome [1]. The clinical presentation can be varied and not the same in all cases. somatic anomalies can present in some cases and absent in others. The inheritance of the forms of 46XX gonadal dysgenesis not associated with somatic malformations is commonly autosomal recessive and more common in consanguineous families. These cases present with normal stature without any Turner's phenotype. Several pleiotropic genes may be involved in 46XX gonadal dysgenesis with somatic malformations. After gonadal dysgenesis, the second most common cause of primary amenorrhea is Mayer Rokitansky Kuster Hauser syndrome (MRKHS) which is characterized by congenital absence or hypoplasia of the uterus and upper two‐thirds of the vagina. Females with this syndrome are usually phenotypically and karyotypically normal females with normal developed secondary sexual characteristics and functioning ovaries. MRKHS affects 1 in 4500 newborn girls [2].
The coexistence of gonadal dysgenesis and MRKHS is very rare, though it has been reported in a few cases and appears to be coincidental as not related to any chromosomal abnormalities.
Case report
We report a case of 17-year-old girl who presented to our clinic with primary amenorrhea, short stature, and absence of secondary sexual characteristics. She had a normal neonatal period with normal developmental milestones. No history of trauma, respiratory symptoms, skin lesions, vaginal discharges, or vaginal bleeding was reported. She was medically and surgically free and doesn’t report any family history of concern, although there is positive consanguinity between her parents. She has a high intellect and a good academic record.
On examination, there were no dysmorphic features, the height was 147, and the weight was around 40 kg. she had normal BP and cardiovascular examination. Breast development, axillary and pubic hair growth staged as ‘Tanner 1’. She had normal external genitalia. Pelvic ultrasound didn’t visualize the uterus or the ovaries (Figure 1). The hormonal assay showed hypogonadotropic hypogonadism status with FSH of 51 and LH of 21. Prolactin and TSH levels were normal.
She had a normal female karyotype 46XX. Brain MRI was normal. Pelvic MRI showed the absence of the uterus, cervix, vagina which had confirmed the diagnosis of Mullerian duct agenesis (Mayer- Rokitansky-Kuster-Hauser syndrome) with bilateral ovarian dysgenesis (Figure 3). Urinary tract abnormalities were excluded as well by MRI. Bone mineral density was found to be less than expected for the patient's age on a BMD scan. Bone age by x-ray (skeletal age) was 14 years with a marked delay in bone maturation (Figure 2).
The echo has been done with normal results. She was also referred to ENT for audiology evaluation which was reported as normal. The patient was started on hormone replacement therapy by conjugated estrogen and was given calcium and vitamin D supplement as well. A few months later the patient was reassessed and found to have an improvement in her height which became 150 cm. Secondary sexual characteristics staged as Tanner 2.
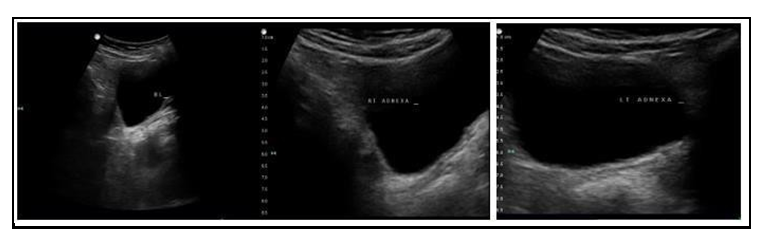
Figure 1: Ultrasound showed absence of uterus and ovaries
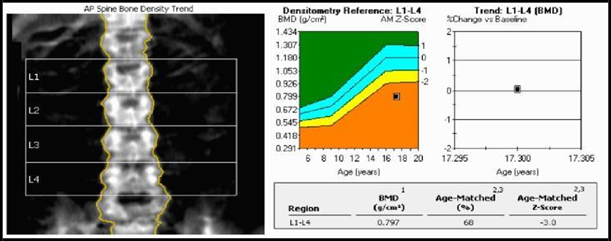
Figure 2: Bone Mineral Density Scan
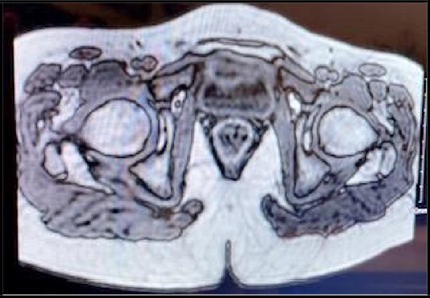
Figure 3: MRI sagittal plane cut showing absence of uterus and ovaries
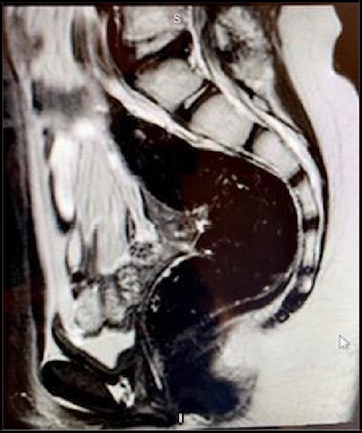
Figure 4: MRI axial plane cut showing bladder and rectum without uterus in-between
Discussion
Mayer Rokitansky Kuster Hauser syndrome is characterized by congenital absence of Mullerian duct derivatives in phenotypically normal females having 46XX chromosomes. The prevalence has been reported as 1 in 4500 female births [3]. It is divided by researcher into 2 types:
- Typical syndrome is characterized by the absence of both the vagina and uterus, leaving only symmetric uterine remnants, normal fallopian tubes, and ovaries.
- Atypical syndrome which shows asymmetric or absent uterine remnants, hypoplasia, or aplasia of one or both fallopian tubes, and frequently associated anomalies [4].
In 40 % of cases, upper urinary tract malformations are found including unilateral renal agenesis, renal ectopia/hypoplasia, horseshoe kidney, and hydronephrosis. Other malformations reported are Skeletal and cardiac anomalies, auditory defects, and anorectal malformations. Screening for these anomalies by ultrasound and MRI and bone scan is mandatory for all cases of MRKH syndrome. Laparoscopy can be done in some cases for final confirmation [3].
In our case, the diagnosis of typical MRKH was made based on pelvic ultrasound, MRI, and we excluded other anomalies by MRI findings, bone scan, cardiac and auditory evaluation. The exact etiology of MRKH syndrome remains largely unknown although many hypotheses have been speculated. Changes in several genes involved during embryological development during the sixth and seventh week of gestation can give rise to the abnormalities seen in the MRKH syndrome [5]. The increased number of cases in familial aggregates raises the genetic cause hypothesis [6]. Estrogens and anti-Mullerian hormones are playing a role in Mullerian duct development. During embryonic development, the existence of activating mutations of either the gene for the anti-Mullerian hormone or the gene for the anti-Mullerian hormone receptor and the lack of estrogen receptors has been hypothesized to cause MRKH syndrome [4].
Gonadal dysgenesis with a female phenotype is defined as the absence or incomplete development of the ovaries and is the most common cause of primary amenorrhea and absent secondary sexual characteristics. In the literature, it was found in different karyotypes like 45XO, 45X/46XX, 45X/46X, dic(X), 46XX, and 46XY [7]. Our case is 46XX which is a relatively rare form of gonadal dysgenesis. The underlying etiology of ovarian dysgenesis is still unknown although several genes have been implicated, including homozygous or compound heterozygous inactivating mutations of follicle-stimulating hormone receptors, and mutations in BMP15 and NR5A1 [8].
The coexistence of Mayer Rokitansky Kuster Hauser syndrome and gonadal dysgenesis has been reported in the literature but is extremely rare. A literature review found 29 reported cases of the concomitant presence of these two abnormalities (Table 1).
Table 1: Summary of published cases of coexisting gonadal dysgenesis and MRKHS
|
Case |
Author |
Year |
Age at presentation (years) |
Karyotype |
Uterus |
Ovaries |
Fallopian tubes |
|
1 |
Arnab Nandy et al. |
2019 |
12 |
46 XX |
Hypoplastic |
Agenetic |
Absent |
|
2 |
Ioris et al. |
2019 |
17 |
46 XX |
Rudimentary Uterine with cervix and normal vagina |
Dysgenetic |
Normal |
|
3 |
Manne C et al. |
2016 |
20 |
46XX |
Absent |
Agenetic |
NR |
|
4 |
Meena et al. |
2016 |
15 |
45X/46XX |
Absent |
Agenetic |
NR |
|
5 |
Białka et al. |
2016 |
17 |
46, X/X(q10) |
Hypoplastic |
Dysgenetic |
NR |
|
6 |
Bhandari and Chaudhary |
2015 |
17 |
46XX |
Absent |
Agenetic |
NR |
|
7 |
Kebaili et al. |
2013 |
21 |
46XX |
Absent |
Agenetic |
Absent |
|
8 |
Viral et al. |
2013 |
21 |
46XX |
Absent |
Agenetic |
Absent |
|
9 |
Bousfiha et al. |
2010 |
19 |
46XX |
Absent |
Dysgenetic |
Absent |
|
10 |
Tatar et al. |
2009 |
2 sisters (34 and 23) |
46XX |
Hypoplastic |
Agenetic |
Hypoplastic |
|
11 |
Zaman and Nisar |
2009 |
2 sisters (22 and 13) |
46XX |
One absent, other rudimentary |
Dysgenetic |
Hypoplastic |
|
12 |
Güvan et al. |
2008 |
17 |
45,X/46,X delX (p11.21) |
Absent |
Agenetic |
NR |
|
13 |
Kumar et al. |
2007 |
18 |
46XX |
Absent |
Right side, Agenetic |
NR |
|
14 |
Colombani et al. |
2007 |
15 |
46XX |
Absent |
Dysgenetic |
Normal |
|
15 |
Marrakchi et al. |
2004 |
19 |
46XX |
Absent |
Dysgenetic |
Normal |
|
16 |
Plevraki et al. |
2004 |
6 patients |
46XX with testis specific protein 1‐Y linked gene (in Patient 1 and 4) |
Patient 1: hypoplastic uterus with symmetrical uterine buds, with no endometrium Patient 6: uterus, symmetrical hypoplastic |
Patient 1: left side, Agenetic Patient 6: Agenetic |
Patient 1: left fallopian tube, absent Patient 6: both fallopian tubes were symmetric, but hypoplastic |
|
17 |
Kaya et al. |
2003 |
17 |
46XX |
Absent |
Left Agenetic |
Right, normal Left, hypoplastic |
|
18 |
Aydos et al. |
2003 |
19 |
46,X,del(X)(pter‐‐ > q22:) |
Rudimentary |
Agenetic |
NR |
|
19 |
Mégarbané et al. |
2003 |
2 sisters |
46XX |
Hypoplasia |
Dysgenetic |
Hypoplastic |
|
20 |
Gorgojo et al. |
2002 |
17 |
46XX |
Absent |
Agenetic |
Absent |
|
21 |
Ting and Chang |
2002 |
22 |
45X/46X, del(X) (p22.22) |
Absent |
Dysgenetic |
Rudimentary |
|
22 |
Güitrón‐Cantú et al. |
1999 |
19 |
45,X/46,Xdic(X) |
Absent |
Agenetic |
Normal |
|
23 |
Oyer et al. |
1994 |
Neonate |
46XX |
Defects in Müllerian derivatives |
Agenetic |
NR |
|
24 |
Albelardo et al. |
1999 |
19 |
45x/46,xdic(x) |
Absent |
Agenesis |
Normal |
|
25 |
Aughton |
1993 |
NA |
46XX |
Absent |
Dysgenetic |
Absent |
|
26 |
Alper et al. |
1985 |
16 |
NA |
Absent |
Dysgenetic |
NR |
|
27 |
Al‐Awadi et al. |
1985 |
2 sisters (18 and 16) |
46XX |
Hypoplastic |
One agenetic, other dysgenetic |
One absent, other hypoplastic |
|
28 |
De Leon et al. |
1984 |
NA |
46, X,i(Xq) |
Absent |
Agenetic |
NR |
|
29 |
Levinson et al. |
1976 |
17 |
46XX |
Absent |
Agenetic |
Absent |
* MRKUS: Mayer Rokitansky Kuster Hauser syndrome, NR: not reported.
The exact genetic mechanisms that underlie the association of Mayer Rokitansky Küster Hauser syndrome with 46XX gonadal dysgenesis are not known [9]. In 1976, Levinson et al. reported the first case of MRHKS in a patient with bilateral gonadal absence and was associated with left sided double ureter [10]. The age of presentation of most of the reported cases was adult age, like our case. Some of the cases had hypoplastic or rudimentary uterus (6 cases). Urological and skeletal abnormalities were reported in a few cases [1,4,10-16]. In our case, we excluded any associated abnormalities.
The presence of these two conditions will compromise the patient’s chances to have her biological children. The only treatment modality will be the use of hormonal replacement therapy to trigger and maintain secondary sexual characteristics and to prevent osteoporosis.
Conclusion
The association between Mullerian duct anomalies (Mayer Rokitansky Kuster Hauser syndrome) and gonadal dysgenesis is a rare event and appears to be a coincidental association and not related to any genetic abnormality. The condition will not only compromise the patient’s fertility potential but also will have a great psychological and social impact on her life.
References
- Manne S, Veeraabhinav CH, Jetti M, Himabindu Y, Donthu K, et al. (2016) A rare case of 46,XX gonadal dysgenesis and Mayer‐ Rokitansky‐Kuster‐Hauser syndrome. J Hum Reprod Sci. 9(4): 263– 266.
- Folch M, Pigem I, Konje JC (2000) Mullerian agenesis: Etiology, diagnosis, and management. Obstet Gynecol Surv. 55(10): 644– 649.
- Rampone B, Filippeschi M, Di Martino M, Marrelli D, Pedrazzani C, et al. (2008) Mayer‑Rokitansky‑Kuster‑Hauser syndrome presenting as vaginal atresia: Report of two cases. G Chir. 29(4): 165‑7.
- Gorgojo JJ, Almodóvar F, López E, Donnay S (2002) Gonadal agenesis 46,XX associated with the a typical form of Rokitansky syndrome. Fertil Steril. 77(1): 185‑7.
- Sharma S, Aggarwal N, Kumar S, Negi A, Azad JR, et al. (2006) Atypical Mayer‑Rokitansky‑Kuster‑Hauser syndrome with scoliosis, renal and anorectal malformation‑case report. Indian J Radiol Imaging. 16(4): 809‑812.
- Morcel K, Camborieux L, Programme de Recherchessur les Aplasies Müllériennes, Guerrier D (2007) Mayer‑Rokitansky‑Kuster‑Hauser (MRKH) syndrome. Orphanet J Rare Dis. 2: 13.
- mäki K (1994) The genetics of XX gonadal dysgenesis. Am J Hum Genet. 54(5): 844– 851.
- Kebaili S, Chaabane K, Mnif MF, Kamoun M, Kacem FH, et al. (2013) Gonadal dysgenesis and the Mayer‐Rokitansky‐Kuster‐ Hauser Syndrome in a girl with a 46, XX karyotype: A case report and review of literature. Indian J Endocrinol Metab. 17(3): 505– 508.
- Kisu I, Ono A, Lijma T, Katayama M, Iura A, et al. (2019) Mayer‐ Rokitansky‐Küster‐Hauser syndrome with a uterine cervix and normal vagina associated with gonadal dysgenesis in a 46,XX female. Journal of Obstetrics and Gynaecology Research. 45(7): 1386-1390.
- Levinson G, Zárate A, Guzmán-Toledano R, Canales ES, Jiménez M (1976) An XX female with sexual infantilism, absent gonads, and lack of Müllerian ducts. J Med Genet. 13(1): 68–9.
- Güven A, Kara N, Sağlam Y, Güneş S, Okten G (2008) The Mayer-Rokitansky-Kuster-Hauser and gonadal dysgenesis anomaly in a girl with 45, X/46, X, del (X)(p11.21). Am J Med Genet A. 146A(1): 128–131.
- Kumar A, Mishra S, Dogra PN (2007) Management of an unusual case of atypical Mayer-Rokitansky-Kuster-Hauser syndrome, with unilateral gonadal agenesis, solitary ectopic pelvic kidney, and pelviureteric junction obstruction. Int Urogynecol J Pelvic Floor Dysfunct. 18(7): 823–5.
- Plevraki E, Kita M, Goulis DG, Hatzisevastou-Loukidou H, Lambropoulos AF, et al. (2004) Bilateral ovarian agenesis and the presence of the testis-specific protein 1-Y-linked gene: Two new features of Mayer-Rokitansky-Küster-Hauser syndrome. Fertil Steril. 81(3): 689–92.
- Kaya H, Sezik M, Ozkaya O, Köse SA (2003) Mayer-Rokitansky- Küster-Hauser syndrome associated with unilateral gonadal agenesis. A case report. J Reprod Med. 48(11): 902–4.
- Aydos S, Tükün A, Bökesoy I (2003) Gonadal dysgenesis and the Mayer-Rokitansky-Kuster-Hauser syndrome in a girl with 46, X, del (X)(pter – >q22:) Arch Gynecol Obstet. 267(3): 173–4.
- Ting TC, Chang SP (2002) Coexistence of gonadal dysgenesis and Mullerian agenesis with two mosaic cell lines 45, X/46, X, del (X)(p22.2). Zhonghua Yi Xue Za Zhi (Taipei). 65(9): 450–2.