


Lamyaa Al Sa’idi1, *, Nasser Al Busaidi2, Muna Al Bimani3
1Department of medicine, Suhar Hospital, Sultanate of Oman
2Department of Chest medicine, Royal Hospital, Sultanate of Oman 3Genetic Center of Oman, Royal Hospital, Sultanate of Oman
*Corresponding Author: Lamyaa Al Sa’idi, Department of medicine, Suhar Hospital, Sultanate of Oman
Abstract:
Background: Cystic Fibrosis is an autosomal recessive disease that affects mainly the respiratory system causing thickened secretions. It is caused by different mutations that affecting the CF transmembrane conduction regulator (CFTR) protein.
Case summary: two Omani siblings were assessed in the respiratory clinic due to respiratory symptoms in terms of chronic cough. Both had recurrent admission initially for pneumonia. CT chest showed bronchiectasis changes and sweat chloride test was positive for both. Both underwent CFTR gene sequence analysis which showed a novel mutation Asp192Val which confirm the diagnosis of CF.
Conclusion: Asp192Val is a novel mutation that was found in 2 Omani siblings in which both had recurrent respiratory symptoms with less pancreatic involvement. Therefore, screening for patients with suspected CF should be extended to involve more mutations.
Keywords: Cystic Fibrosis, CF transmembrane conductance regulator, Asp192Val.
Introduction
Cystic Fibrosis is an inherited autosomal recessive disease that can affect different systems including upper and lower respiratory, gastrointestinal, endocrine, and reproductive systems [1-5].CFTR is a complex chloride channel and regulatory protein which maintains the fluid balance in airways [2]. CF is caused by a mutation in CF transmembrane conduction regulator (CFTR) gene which is located on the long arm of chromosome 7 [3,13]. There are six classes of CFTR mutations based on how CFTR protein is affected by the mutations [4- 5]. Delta F508 is the most common mutation of CFTR in Western countries [6,8-9]. A study which was conducted in Oman by Al-kindly including patients from 2000 to 2011 concluded that p.S549R is the most prevalent mutation in Middle Eastern countries including Oman followed by Delta F508[1].
CF is more common in western countries compared to eastern countries [3-4]. The prevalence in Northern Europe estimated to be 1/2500 – 3500 whereas in the Asian population estimated to be 1/31,000 [6-7]. The leading cause for morbidity and mortality in CF is a rapid deterioration of pulmonary function related to thickened secretions that might damage airways [10-12].
Patient 1:
A young girl aged 26 years presented to Royal Hospital, chest clinic in April 2011with recurrent productive cough of greenish sputum since the age of 5 years. She had experienced recurrent pneumonia (1-2 times every year), but no history of pancreatic involvement or chronic diarrhea. She was diagnosed to have Barter syndrome as well. Her parents are first-degree cousins. Her elder brother died at the age of 5 years after repeated pneumonia. On physical examination she weighed 55 kg, her height was 161 cm, her body mass index was 22.1. She had no clubbed digits and her chest was clear. The heart and abdomen were normal. On laboratory examination, her routine investigations showed a white cell count of 7.1 × 109/L, a C reactive protein concentration of 12.9 mg/L. Elastase stool found to be > 500 ug/g of feces. Sputum culture grew Moraxella catarrhalis and Staphylococcus aureus. Sweat chloride test resulted as 72 mmol/L (Diagnostic > 60 mmol/L) and sweat conductivity was 100 mmol/L (suggestive > 80 mmol/L). Pulmonary Function Test was done during the first visit and showed FEV1 1.76 (62 %), FVC 2.35 (72 %), FEV1/FVC 75 %, TLC 3.12 (64 %), DLCO 18.6 (72 %), with no reversibility. High-Resolution CT showed bilaterally upper lobe bronchiectasis more in the right side with bronchialwall thickening (Figure 1).
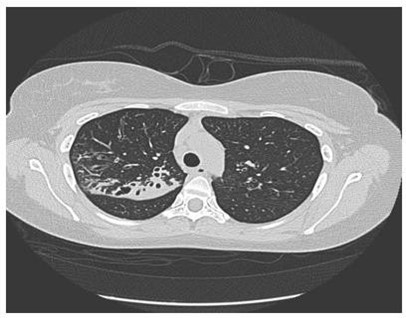
Figure 1: HRCT showed bilateral upper lobe bronchiectasis more in right side.
CFTR gene sequence analysis
The Genomic DNA of this patient was extracted by the magic STAR– Hamilton automated machine.48 CFTR amplicons (300-400pb) targeting all coding exons, Selected intronic regions (intron7,12, and 22) and part of the promoter region was amplified by Multiplicom's CFTR MASTR DXassay. Next-generation sequencing was performed on the illumine MI seq and results were analyzed by Sex Next. Case result was confirmed by bidirectional Sanger sequencing for exon 5 and analyzed by seqpilot (Figure 2), This patient is found homozygous for likely pathogenic mutation NM_000492.3 (CFTR): c.575A>T, p. Asp192Val, traditional name D192V .it is in the ABC transporter type 1, transmembrane domain (via InterPro) involves the alteration of a conserved nucleotide. It is predicted to have a damaging outcome in Mutation Taster, SIFT, and Polyphen tools. However, there is no functional study on it, and it has not been reported in affected individuals via publications and/or reputable databases/clinical diagnostic laboratories. Because of the absence of clinical information and the lack of functional studies, National Center for Biotechnology Information (NCBI) classified this variant as "Variant of Uncertain Significance (VUS) [15]. A segregation study was done in our lab for this patient's parents, and they were found heterozygous for the same variant which confirms the pathogenicity of this mutation, so the genetic study has confirmed the diagnosis of Cystic Fibrosis of this patient.
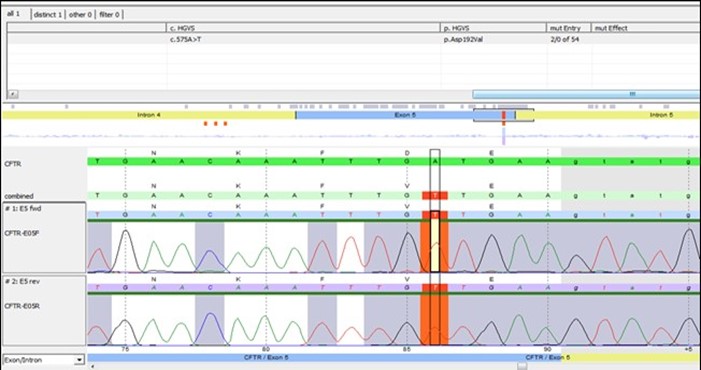
Figure 2: Sequence analysis by Seq Next showing the substitution mutation clearly.
Treatment, outcome and follow up
The patient was started on chest physiotherapy, airway clearance measures including dornase alpha, azithromycin as anti-inflammatory, and Seretide Diskus. The patient remains stable throughout and did not require admission. She is under follow up in the chest clinic every 3 months.
Patient 2:
A middle-aged gentleman, 31 years presented to Royal Hospital, chest clinic in April 2011, with a history of recurrent productive cough. His younger sister was diagnosed with CF and referred to the clinic to rule out CF. He was diagnosed to have barter syndrome. On physical examination, He weighed 69 kg, height 176 cm and body mass index were 22 kg/m2. He had no clubbed fingers and his chest had bilateral basal coarse crackles. His cardiovascular system was normal, andhisabdomen was normal as well. On laboratory examination, sputum culture grew Pseudomonas aeruginosa and staphylococcus aureus. His sweat chloride test resulted as 86 mmol/L (Diagnostic > 60 mmol/L) and sweat conductivity resulted as 113 mmol/L (suggestive > 80 mmol/L). A full pulmonary function test was done the first visit and showed FEV1 3.43 (81 %), FVC 4.23 (85 %), FEV1/FVC 81 %, TLC 5.39 (80 %). Computed Tomography was done and showed dilated bronchi with peribranchial thickening involving the right upper, middle lobe, and left upper lobe. There were multiple areas of branching density seen within the involved lobes with the tree inBudd pattern indicating endobronchial spread. Areas of segmental peripheral consolidation seen within posterior segment of right upper lobe, medial lingual, and more marked in the rightmiddle lobe (Figure 3). The patient underwent bronchoscopy, found to have inflamed mucosa in RML, and lingual with thick purulent sputum. TB culture grew Mycobacterium avium- intracellular.
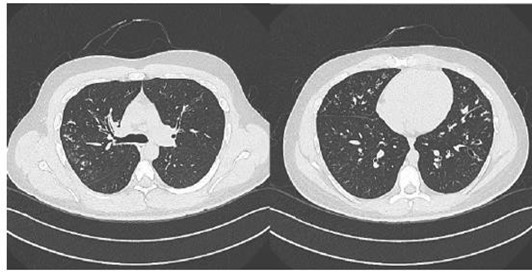
Figure 3: CT chest showing multiple areas of branching density with a tree in Budd pattern.
CFTR gene sequence analysis
Exon 5 of this patient was amplified by long-range PCR program see (table1) by using specific primers see (table2) and 5X HOT FIREPol®Blend Master Mix. The PCR product was visualized using Caliper. Then, Sanger sequencing was performed on the 3130xl genetic analyzer. The resultwas analyzed by seqpilot. This patient is found homozygous for the same likely pathogenic mutation NM_000492.3 (CFTR): c.575A>T, p. Asp192Val, traditional name D192V.
Table1: long range PCR program
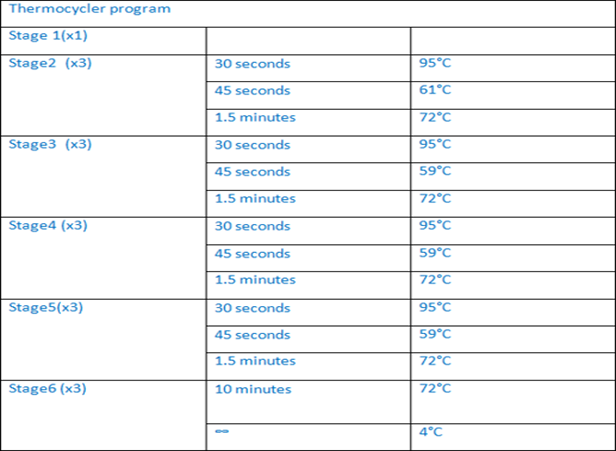
Table2: Exon 5 tag primers
Treatment, outcome and follow up
The patient was treated for MAC for 18 months’ course. He was started on azithromycin as an anti-inflammatory. Also, He was started on pancreatin capsule 2000 Iu three times a day and dornase alpha nebulizing solution 1.25 mg once daily. In addition, he was treated for allergicrhinitis with fluticasone propionate nasal spray 50 mcg twice a day. The patient is under regular follow-up every 3 months in the chest clinic.
Discussion
CF is a genetic multisystem disease that can be caused by different mutations affecting CFTR protein [1,4]. There are six classes of mutations and more than 2000 mutations in the CFTR gene [2]. These mutations were grouped based on disruption of CFTR protein production and function [3]. Nonsense and spliced mutations in Class I can result in a premature stop of CFTR protein production which can lead to little or no expression of functional CFTR protein [4-5]. Class II mutations affect the processing of CFTR protein in which there is the deletion of amino acids or incorporation of incorrect amino acids in CFTR protein. An example of these mutations is Delta F508del in which there is a deletion of three nucleotides from exon 10 that result in loss of the amino acid phenylalanine at position 5081. Patients who are homozygous for both class I and II tend to develop severe disease [13]. Class III mutations usually locking the gate in a closed position, so preventing the chloride to flow through the channel despite the expression of the full protein in the apical plasma membrane of epithelial cells [1-2]. Class IV mutations tend to reduce the permeability of chloride [14]. Mutations in class V can lead to reducing the expression of the normalCFTR protein at the cell surface while in class VI mutations, the protein has a short residence time at the apical plasma membrane [14]. Individuals with mutations IV-VI usually have a mild form of the disease [10-12,14]. In this case report, a novel mutation was found in two siblings of the same family which is Asp192Val. In this mutation, there is a substation of amino acid aspartate for valine at position 192. Both patients have mainly respiratory involvement with less pancreatic involvement. They experience respiratory symptoms in terms of the main cough with sputum production. Both patients have changes in their CT chest which more going with bronchiectasis changes.
The determination of this novel mutation has shed the light on the importance of extending the screening programs for determining CF mutations. Early detection of such mutations can help indetermining the severity of the disease and further planning the appropriate care of those patients in all aspects especially in the context of the new cystic fibrosis transmembrane conductance regulator (CFTR) - modulating drugs.
Conclusion
In conclusion, Asp192Val is a novel mutation that was found in two siblings of the same family inOman. Those patients are experiencing respiratory symptoms with changes in their imaging. Both are under the care and follow-up of respiratory team in royal hospital, Oman.
References
- Al-Kindy H, Ouhtit A, Al-Salmi Q, Al-Bimani M, Al-Nabhani M, et al. (2014) Novel Mutation inthe CFTR Gene of Cystic Fibrosis Patients in Oman. J Mol Biomark Diagn 5(2): 168.
- Wang YQ, Hao CL, Jiang WJ, Lu YH, Sun HQ, et al. (2019) c.753_754delAG, a novel CFTR mutation found in a Chinese patient with cystic fibrosis: A case report and review of the literature. World J Clin Cases. 7(15): 2110-2119
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, et al. (1989) Identification ofcystic fibrosis gene: genetic analysis. Science 245(4922): 1073-1080.
- Elborn JS (2016) Cystic fibrosis. Lancet. 388(10059): 2519–2531.
- O'Sullivan BP, Freedman SD (2009) Cystic fibrosis. Lancet. 373(9678): 1891–904.
- Filbrun AG, Lahiri T, Ren CL. (2016) Introduction and Epidemiology of Cystic Fibrosis. Handbook of Cystic Fibrosis. Cham: Springer International Publishing. p. 1–4.
- Bobadilla JL, Macek M Jr, Fine JP, Farrell PM (2002) Cystic fibrosis: a worldwide analysis of CFTR mutations--correlation with incidence data and application to screening. Hum Mutat. 19(6): 575-606.
- Ratjen F, Bell SC, Rowe SM, Goss CH, Quittner AL, et al. (2015) Cystic fibrosis. Nat Rev DisPrimers. 1: 15010.
- el-Harith EA, Dörk T, Stuhrmann M, Abu-Srair H, al-Shahri A, et al. (1997) Novel and characteristic CFTR mutations in Saudi Arab children with severe cystic fibrosis. J Med Genet. 34(12): 996-999.
- Riordan JR, Rommens JM, Kerem B, Alon N, Rozmahel R, et al. (1989) Identification of the cystic fibrosis gene: cloning and characterization of complementary DNA. Science. 245(4922): 1066-1073
- Kerem B, Rommens JM, Buchanan JA, Markiewicz D, Cox TK, et al. (1989) Identification of the cystic fibrosis gene: genetic analysis. Science. 245(4922): 1073-1080.
- Davis PB (2006) Cystic fibrosis since 1938. Am J Respir Crit Care Med. 173(5): 475-482
- Chami H, Arbid SA, Badra R, Farra C (2017) A novel cystic fibrosis gene mutation c.2490insT in aPalestinian patient: A case report and review of the literature. Ann Thorac Med. 12(4): 290- 293.
- Mohseni M, Razzaghmanesh M, Mehr EP, Zare H, Beheshtian M, et al. (2016) Novel CFTR Mutations in Two Iranian Families with SevereCystic Fibrosis. Iranian Biomedical Journal 20(4): 201-206.
- https://www.ncbi.nlm.nih.gov/clinvar/variation/633172/