


Shamim J1*, Pillai C2, Stranberg A3, Coon JI4, Richardson CJ4
1Pediatric Resident at UTMB, University of Texas Medical Branch (UTMB) at Galveston, TX.
2Division of Endocrinology, University of Texas Medical Branch (UTMB) at Galveston, TX.
3Neonatology Fellow at UTMB, University of Texas Medical Branch (UTMB) at Galveston, TX.
4Division of Neonatology, University of Texas Medical Branch (UTMB) at Galveston, TX.
*Corresponding Author: Shamim J, Pediatric Resident at UTMB, University of Texas Medical Branch (UTMB) at Galveston, TX.
Abstract
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder of adrenal steroidogenesis. Clinical manifestations depend on the degree of gene deletion or mutation. Classic CAH presents at birth with salt wasting and females with ambiguous genitalia. Non-classic CAH presents later in life with androgen excess.
This case was atypical. Although our patient had non-classic CAH with sky-high testosterone levels, our lack of significant concern in the beginning was primarily due to the reassuringly robust cortisol levels, which made adrenal insufficiency seem unlikely, coupled with the absence of results for 17-OHP in the first week of life and normal adrenals on imaging. Rapid exome sequencing yielded non-diagnostic results. The CAH panel showed a mutation in the outpatient.
Keywords: Ambiguous genitalia, non-classic congenital adrenal hyperplasia, salt wasting, 21-hydroxylase deficiency, genetic testing, high testosterone.
Introduction
Congenital adrenal hyperplasia (CAH) is an autosomal recessive disorder of adrenal steroidogenesis presenting with salt wasting and ambiguous genitalia in females in the neonatal period or infancy [1]. Clinical manifestations depend on degree of gene deletion or mutation varying from silent mutation carrier to salt wasting form with virilization of genitalia. CAH is a cannot miss diagnosis. Early diagnosis and treatment can prevent adrenal crisis and death [2,3]. Classic CAH presents at birth with salt wasting and females with ambiguous genitalia [4]. Non-classic CAH presents later in life with androgen excess5. We present a case where the diagnosis was unclear during the initial evaluation.
Case Description
Our patient is a term baby, born at 39 weeks of gestation to a primigravida mom. Birth weight was 2.990 kg. APGARs were 7 & 8 at 1 & 5 minutes of life respectively. Mom did not receive prenatal care and there were no documented complications during pregnancy, labor, and delivery. The newborn was admitted to the NICU with respiratory distress due to transient tachypnea of the newborn requiring oxygen support. The baby was also noted to have ambiguous genitalia (clitoromegaly with labial fusion- Figure 1) and no palpable gonads on the exam.
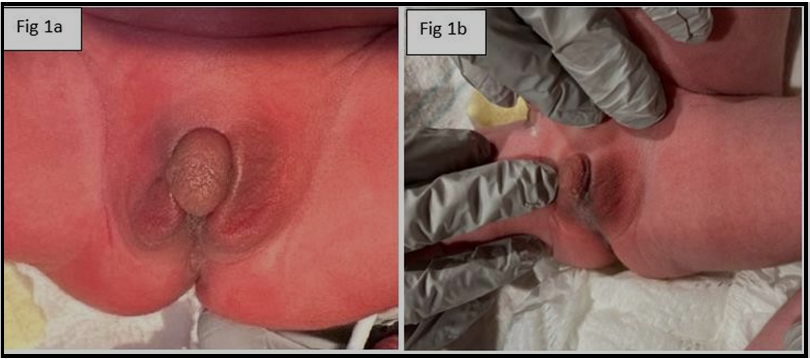
Figure 1: ambiguous genitalia (clitoromegaly with labial fusion) and no palpable gonads on the exam.
Mom denied recreational drug use or over-the-counter supplements during pregnancy besides prenatal vitamins. There was no family history of ambiguous genitalia. Pelvic ultrasound showed a uterus, left ovary (right ovary not visualized), dilated cervix, and upper vagina concerning for imperforate hymen. LH, and FSH both were <0.20 mIU/ml (normal). Testosterone was 1860 ng/dL (16-64 ng/dL in newborn girls and 75-400ng/dL in newborn boys) in the first 24 hours of life. 17-Hydroxyprogesterone (17-OHP) on day of life (DOL) 1 was 2552 ng/dL (7-106 ng/dL). The testosterone level was>2160 ng/dL on DOL 3. An Adrenocorticotropic hormone (ACTH) stimulation test showed a peak cortisol of 80.5 ug/dL with a baseline of 29.8 ug/dL. 17-OHP was 7305.5 ng/dL. Adrenal glands were not enlarged on imaging. MRI of the abdomen and pelvis showed a uterus and no masses or cysts. Anti-mullerian hormone 0.258 ng/mL (normal). Our diagnosis was unclear until the patient started salt-wasting on DOL 7 (Graph 1,2). At that time the baby was hypotensive 75/50 mm Hg with low mean arterial pressure (MAP) 33-35 mm Hg, tachycardiac with a heart rate of 80-190/minute, poor feeding, lethargic with concern for abdominal distension. We gave a normal saline bolus of 10 ml/kg, but the baby did not respond. We gave packed red blood cells at 20 ml/kg without any improvement. We gave a stress dose of hydrocortisone (100 mg/m2) and the baby responded well. Per routine NICU management, after initial stabilization, we obtained baseline labs and septic work to rule out infection vs adrenal crisis where the basal metabolic panel indicated salt wasting as evidenced by serum sodium 130 (133-145 mmol/L) and serum potassium 6.7 (3.0-6.0 mmol/L) from a non-hemolyzed sample. Electrolyte abnormality resolved with steroid treatment and oral sodium chloride supplement (Figure 2 and 3).
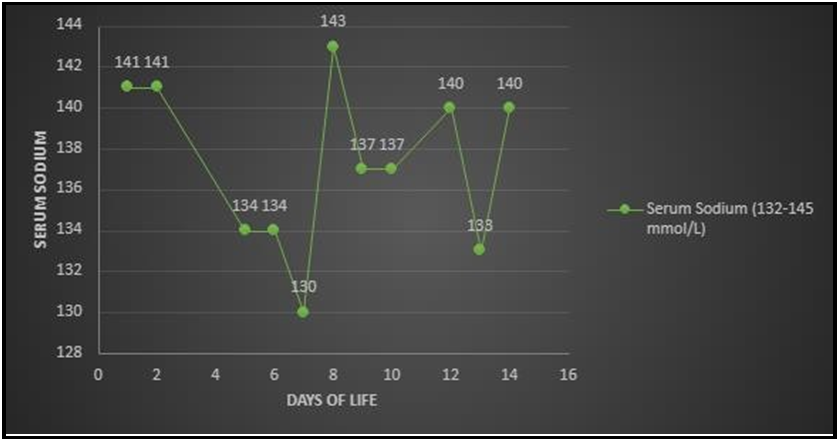
Figure 2: Serum sodium levels within first few days of life.
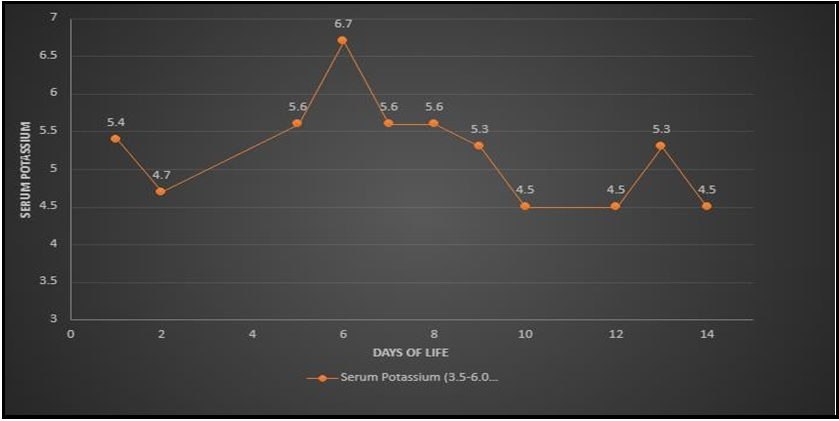
Figure 3: Serum potassium levels within first few days of life.
Repeated testosterone was 4 ng/dL. Newborn screens 1 &2 were flagged for 21 hydroxylase deficiency. Fluorescence in situ hybridization (FISH) for the SRY gene (sex-determining region on Y-chromosome) was negative. The karyotype was XX. Rapid whole exome sequencing (WES-DUO maternal) was negative. She was started on fludrocortisone (0.1 mg twice daily) and prednisolone (0.75 mg once daily) along with oral salt supplementation and responded to treatment. Multiplex Ligation-Dependent Probe Amplification (MPLA) showed the presence of two copies of the CYP21A2 gene and two copies of inactive CYP21A1P pseudogene, while DNA sequencing identified pathogenic homozygous variant c.293-13C>G which confirmed the diagnosis of CAH. Parents received regular updates during multidisciplinary team meetings. Parents were educated about the importance of compliance with medication, and stress dosing of steroids at the onset of illness before being discharged. Our patient was discharged home with close follow-ups with endocrine, genetics, urology, and primary care. An informed written consent was obtained from parents before writing this case. Figure 1 taken from the patient’s chart with parental consent.
Discussion
Elevated 17-OHP levels were in the 2000s range, indicative of a closer resemblance to non-classic CAH where 17-OHP is <10,000 ng/dL6. With our limited laboratory data, the case was perplexing until the salt-wasting became apparent. ACTH stimulation test is instrumental in diagnosis confirmation as non-classic CAH has lower peak cortisol [7]. Our lack of significant concern at that time was primarily due to the reassuringly robust cortisol levels, which made adrenal insufficiency seem unlikely, coupled with the absence of results for 17-OHP in the first week of life. Similar results were observed in a study conducted at Dankook University Hospital in Korea where ACTH stimulation test resulted in a proportionately appropriate cortisol response in a non-classic CAH patient [8].
Since karyotype results aren't immediately available, gender assignment team recommends to conduct imaging of the abdomen and pelvis to promptly confirm the presence or absence of Müllerian structures and gonads. This allows for the determination of the patient as either a virilized female male (46, XX) in the presence of Müllerian structures or an under virilized male (46, XY) in their absence [9]. Our patient had female internal reproductive organs and we disclosed gender in a family meeting after obtaining karyotype as well as imaging.
Although our patient had non-classic CAH, adrenals were not enlarged on MRI. This was contrary to previous studies where adrenal enlargement correlated with hormone levels [10].
We were initially concerned about the sky-high testosterone level of 2160 ng/dL as this is unusually high even for salt wasting CAH (Table 1). Testosterone producing tumors in the neonate or mother were high on the differential.
Table 1: 17-OHP and testosterone levels before and after treatment.
|
Test |
Before Treatment (ng/dL) |
After Treatment (ng/dL) |
|
17-OHP (7 -106 ng/dL) |
2552 (DOL 0), 2552.2 (DOL 2) |
7305.55 (DOL 14), 261.58 (DOL 22) |
|
Testosterone (16 - 64 ng/dL) |
1860 (DOL 0), >2160 (DOL 2), 1000 (DOL 5) |
4.0 (DOL 22) |
CYP21A2 gene encoding 21-hydroxylase is on chromosome 6p21 [10]. Rapid exome sequencing yielded nondiagnostic results which did not rule out CAH due to pseudogene abnormality (a non-coding DNA segment that regulates gene function). MPLA showed the presence of two copies of the CYP21A2 gene and two copies of inactive CYP21A1P pseudogene, while DNA sequencing identified pathogenic homozygous variant c.293-13C>G which confirmed the diagnosis of CAH. The CAH panel showed a mutation in the outpatient. Pathogenic variants are more commonly seen with non- classic CAH [11]. MPLA is the most effective genotyping modality in patients with CAH due to the presence of pseudogene and pathogenic variants [12].
Conclusion
An adrenal crisis is an endocrine emergency, and it is crucial to identify salt-wasting CAH before the onset of the adrenal crisis. Salt- wasting CAH can present with a very high testosterone levels and paradoxically robust cortisol levels. Physicians cannot rely on ACTH stimulation test to exclude the diagnosis.
Conflict of Interest: All the authors declare that there is no conflict of interest.
Source of Funding: none.
References
- Vats P, Dabas A, Jain V, Seth A, Yadav S, et. al. (2020) Newborn Screening and Diagnosis of Infants with Congenital Adrenal Hyperplasia. Indian Pediatr. 57(1): 49-55.
- Khanal D, Mandal D, Phuyal R, Adhikari U (2020) Congenital Adrenal Hyperplasia with Salt Wasting Crisis: A Case Report. JNMA J Nepal Med Assoc. 58(221): 56-58.
- Gaxiola A, Kumar R, Davis V (2014) Congenital Adrenal Hyperplasia: A Newborn With Ambiguous Genitalia. Consultant.
- Livadas S, Bothou C (2019) Management of the female with non- classical congenital adrenal hyperplasia (NCCAH): a patient- oriented approach. Frontiers in endocrinology. 10: 366.
- Carmina E, Dewailly D, Escobar-Morreale HF, Kelestimur F, Moran C, et al. (2017) Non-classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency revisited: an update with a special focus on adolescent and adult women. Human reproduction update. 23(5): 580-99.
- Gören r, Kocaay p. non-classical congenital adrenal hyperplasia. adrenal diseases in children and adolescents.:43.
- Karachaliou FH, Kafetzi M, Dracopoulou M, Vlachopapadopoulou E, Leka S, Fotinou A, et al. (2016) Cortisol response to adrenocorticotropin testing in non-classical congenital adrenal hyperplasia (NCCAH). Journal of Pediatric Endocrinology and Metabolism. 29(12): 1365-1371.
- Koh JW, Kim GH, Yoo HW, Yu J (2013) Clinical features of congenital adrenal insufficiency including growth patterns and significance of ACTH stimulation test. J Korean Med Sci. 28(11): 1650-6.
- Parisi MA, Ramsdell LA, Burns MW, Carr MC, Grady RE, et al. (2007) Gender Assessment Team: experience with 250 patients over a period of 25 years. Genetics in Medicine. 9(6): 348-57.
- Teixeira SR, Elias PC, Andrade MT, Melo AF, Elias Junior J (2014) The role of imaging in congenital adrenal hyperplasia. Arquivos Brasileiros de Endocrinologia & Metabologia. 58(7): 701-8.
- Falhammar H, Wedell A, Nordenström A (2015) Biochemical and genetic diagnosis of 21-hydroxylase deficiency. Endocrine. 50: 306-14.
- de Carvalho BC (2022) Characterization of the pathogenic variants landscape in 21-hydroxylase deficiency.
- Monteiro A, Pavithran PV, Puthukulangara M, Bhavani N, Nampoothiri S, et al. (2023) Cost-effective genotyping for classical congenital adrenal hyperplasia (CAH) due to 21- hydroxylase deficiency (21-OHD) in resource-poor settings: multiplex ligation probe amplification (MLPA) with/without sequential next-generation sequencing (NGS). Hormones. 22(2): 311-20.