


João Leite MD*, Pedro Manuel Baptista, Miguel Mesquita Neves, Miguel Gomes, Luis Oliveira
Department of Ophthalmology, Centro Hospitalar Universitário do Porto, Porto, Portugal.
*Corresponding Author: João Leite MD, Department of Ophthalmology, Centro Hospitalar Universitário do Porto, Porto, Portugal. ORCID: 0000-0002-7071-2228
Abstract
Ichthyosis is characterized by skin thickening and desquamation, with most cases being hereditary and clinically apparent in the first year of life. Ichthyosis can affect the eyelids (with hyperkeratotic lesions, and eyelash scales, among others) and the cornea (namely by punctate keratitis, recurrent epithelial defects, and corneal perforation).
Hyper-IgE syndrome is a rare primary immunodeficiency that involves both humoral and cellular immune systems. Recurrent skin infections, elevated serum IgE levels, eosinophilia, respiratory tract infections, and chronic eczema characterize it.
We report a case of a child with a clinical diagnosis of Congenital Ichthyosis (Ichthyosis Vulgaris) and Hyper-IgE Syndrome, with an associated bilateral recurrent corneal ulcer history. He was treated successfully with ocular lubrication, and topical corticosteroid tapered slowly.
This case report highlights the importance of a multidisciplinary approach for such a challenging case and demonstrates the preponderance that the ocular surface can assume in rare but incapacitating systemic diseases.
Keywords: Hyper-IgE; Ichthyosis; Ocular diseases; recurrent corneal ulceration; topical corticosteroids.
Introduction
Ichthyosis, also called keratinization disorders or cornification disorders, was first described in 1834 and included a heterogeneous group of diseases. Most cases are inherited and clinically apparent in the first year of life and are characterized by thickening of the skin and scaling [1,2]. Although most patients can be classified based on the mode of inheritance [3], it can also be acquired, sharing the exact characteristics of rough, dry skin with marked scaling and no signs of inflammation [4].
Autosomal dominant ichthyosis Vulgaris and X-linked recessive ichthyosis are the two most common forms in approximately 1/300 and 1/2500 male births, respectively [5]. The untreated state is characterized by visible and symmetrically distributed skin scales (most apparent on the extensor surfaces of the extremities but may also appear on the trunk) and skin xerosis (a prominent feature in most patients with no skin inflammation) [2,5].
Regarding ocular manifestations, ichthyosis can affect the eyelids, causing hyperkeratotic lesions, eyelash scales, madarosis, trichiasis, ciliary retraction, meibomitis, lower eyelid ectropion, abnormal movements of the upper eyelids, and cicatricial lagophthalmia [6]. The conjunctiva usually appears normal [2]; however, due to changes in the eyelids, it can become hyperaemic, thickened (due to chronic exposure), and appear to be chemosis [7]. The cornea may present with recurrent epithelial defects, speckled keratitis, early band-shaped keratopathy, keratoconjunctivitis sicca, deeper subepithelial stromal opacities, leukomas, neovascularization, and the appearance of corneal nerves. Corneal perforation may occur in the most severe cases due to corneal exposure associated with limbic stem cell deficiency [1,2]. Although fundoscopy is essentially normal, nuclear cataracts or other lens opacities can be described; Other findings associated with congenital ichthyosis include coloboma of the iris, choroid and retina, optic neuropathy, and crystalline macular dystrophy [2,6].
Hyper IgE syndrome is a rare primary immunodeficiency that involves the humoral and cellular immune systems [8]. Besides its first description in 1966, the autosomal recessive (associated with loss-of-function mutations in the DOCK 8 gene) and autosomal dominant (associated with signal transducer and activator gene mutations of STAT3 transcript) variants have only been reported at the beginning of the 21st century [8,9].
These two forms are characterized by recurrent skin infections (recurrent staphylococcal abscesses), elevated serum IgE levels, respiratory tract infections, and chronic eczema [8,9,10]. Other manifestations include skeletal, connective tissue (such as scoliosis, osteoporosis, fracture after minor trauma, and extensive hyper joints) and dental abnormalities - in the autosomal dominant form - and susceptibility to viral infections and neurological complications - in the autosomal recessive condition [8,11]. These abnormalities may be accompanied by serological alterations, namely the total elevation of IgE and eosinophilia [12,10].
Case Report
We report the case of a 7-year-old male referred to our Ophthalmology Department after recurrent corneal ulcers. The child had a clinical diagnosis of congenital Ichthyosis (Ichthyosis Vulgaris) owing to intense scaling of the hands, trunk, and face skin during the first year of life. During that year, he was also diagnosed with Hyper- IgE Syndrome after consecutive episodes of recurrent cutaneous abscesses associated with clinically apparent food intolerance in the form of eczematous and urticariform reactions during the introduction of several foods. Serological analyses demonstrated elevated levels of Immunoglobulin E, total and specific, for several antigens. (Tables 1 and 2).
Table 1: Serological values demonstrated a large elevation of serum IgE.
|
Immunoglobulins |
Value obtained |
|
IgA |
103.7 mg/dL |
|
IgG |
805.0 mg/dL |
|
IgM |
82.7 mg/dL |
|
IgE |
13639.0 KU/L |
|
Autoantibodies |
Value obtained |
|
Anti-transglutaminase IgA |
0.2 U/mL |
Table 2: Research on food allergens showed mostly values obtained above the reference values considered normal.
|
Allergy detection |
Value obtained |
Reference value |
|
Milk |
> 100 κUA/L |
< 0.35 |
|
Alpha-lactoalbimune |
75.9 κUA/L |
< 0.35 |
|
Beta-lactoalbimune |
60.8 κUA/L |
< 0.35 |
|
Casein |
> 100 κUA/L |
< 0.35 |
|
Eggwhite |
> 100 κUA/L |
< 0.35 |
|
Egg yolk |
40.6 κUA/L |
< 0.35 |
|
nGal D2 ovalbumine |
57.2 κUA/L |
< 0.35 |
|
nGal D1 ovomucoid |
> 100 κUA/L |
< 0.35 |
|
Wheat |
> 100 κUA/L |
< 0.35 |
|
rTri a 19; Omega-5 gidina |
5.8 κUA/L |
< 0.35 |
|
Peanut |
41.6 κUA/L |
< 0.35 |
|
Hazelnut |
23.3 κUA/L |
< 0.35 |
|
Almond |
28.4 κUA/L |
< 0.35 |
|
Nut |
86.4 κUA/L |
< 0.35 |
|
Soybean |
18.8 κUA/L |
< 0.35 |
|
Whitefish |
> 100 κUA/L |
< 0.35 |
|
Kiwi |
> 100 κUA/L |
< 0.35 |
|
Beef |
15.4 κUA/L |
< 0.35 |
Questioning the personal background, the parents reported that, between the 3rd and 4th years of age, he underwent several cycles of antihistaminic medication accompanied by oral corticosteroid therapy, and the disease progressed, presenting an outbreak-remission pattern. At 4 years of age, it was decided to start chronic daily treatment with oral montelukast 10mg (leukotriene receptor antagonist) and a desensitization scheme for several foods, including cow's milk. Regarding family history, the mother had a history of asthma and angioedema reactions.
At the first consultation in our department, uncorrected visual acuity (UCVA) was 20/25 in each eye, orthotropic for far and near, and with fine stereopsis. The parents reported several periods of red eye and eye pain since the child's first year. They said 4 previous episodes of corneal ulcers (two in each eye) were treated with a short cycle of topical antibiotic and corticosteroid. From the first episode, the child is on ocular lubricating therapy.
Slit lamp examination revealed 2 inferior corneal ulcers (1 in each eye), with a dense and well-delimited opacified ring at the edges, without abscess formations—both ulcers stained with fluorescein. The break-up time was diminished. There was mild symblepharon of the lower conjunctiva in both eyes. There were no other alterations in the anterior segment exam. The intraocular pressure was 15 mmHg in both looks, and bilateral fundoscopy was unremarkable. There was no known history of ocular trauma. Extra-ocular examination showed skin scaling in the trunk, hands, and face (Figure 1).
The diagnosis of mixed corneal ulcers - inflammatory, neurotrophic, and exposure components - was assumed. The decision was to reinforce the ocular lubrication every 2 hours with an association gel of trehalose 3 %, sodium hyaluronate 0,15 %, and carbomer 0,25 % and to add topical hydrocortisone (3,35mg/mL) 5 times a day. Topical hydrocortisone was tapered every 5 days up to once a day. On the 20th day, there was a significant clinical improvement as the infiltrate was milder, and the epithelial defect was almost healed (Figure 2). The child was maintained on ocular lubrication every 2 hours and topical hydrocortisone once a day, and three months after the first visit, the child was asymptomatic with bilateral UCVA of 20/25. Biomicroscopy revealed no signs of ocular inflammation and no evidence of corneal ulcer or significant corneal opacification. There was mild inferior punctate keratitis, and the inferior symblepharon remained unchanged (Figure 3).
The intraocular pressure was 15 mmHg in both eyes. Despite an excellent initial response, the patient is awaiting approval from our hospital to start corticosteroid-sparing therapy with topical 0.05 % cyclosporine.
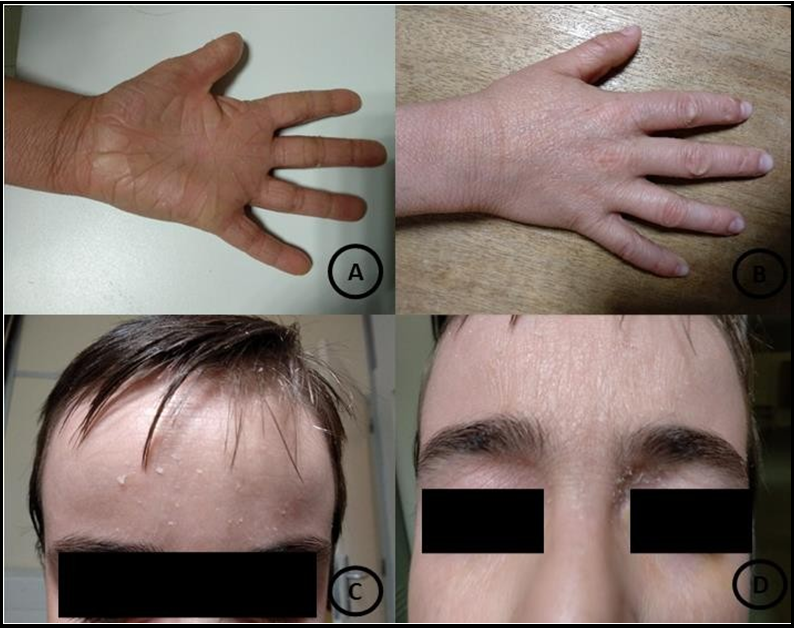
Figure 1: Skin scaling in the trunk, hands, and face.

Figure 2: Biomicroscopy, epithelial defect almost completely healed.
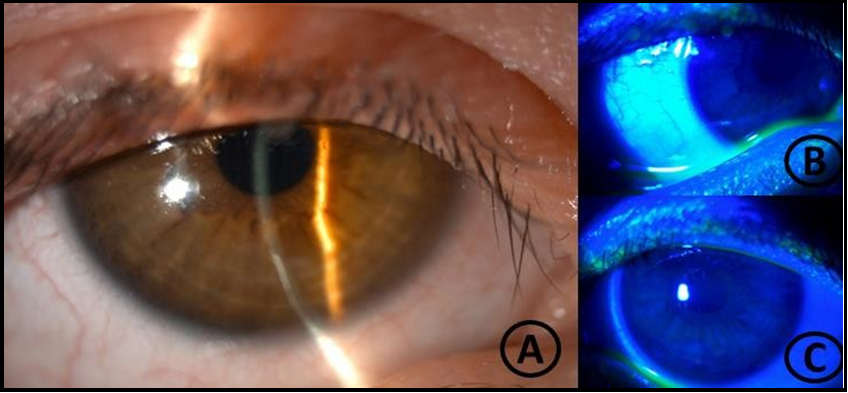
Figure 3: Biomicroscopy revealed inferior punctiform keratitis and symblepharon.
Discussion/Conclusion
Systemic inflammatory diseases, especially those affecting the mucous membranes, can encompass a serious chronic ocular surface problem. The presentation may range from a mild dry eye syndrome to a more severe disease that can lead to recurrent corneal ulcers and long-term conditioning morbidity and may result in a marked loss of visual acuity if not promptly diagnosed and treated. In all cases, a loss of quality of life is associated with permanent ocular discomfort.
In the presented case, there was an association between an inflammatory disease and a congenital condition that aggravates the skin and mucous membranes dryness complicating the whole picture, decreasing life quality since the first years of life and putting in serious risk visual function in the medium and long term.
The approach to such a challenging case must integrate different areas. At the ophthalmological level, it is essential to ensure, first of all, the maintenance of ocular surface homeostasis with efficient lubrication and, not least, to reduce the levels of inflammation in the ocular tissues effectively. We decided to initiate a low-power corticosteroid therapy that proved effective in disease control with no deleterious effects.
The patient had other 4 previous episodes of corneal ulcers (two in each eye), treated with a short cycle of topical antibiotic and corticosteroid. These frequent relapses with good clinical responses to topical corticosteroids support the idea that the patient is dependent on topical corticosteroids to control the ocular manifestations of the disease. Because of that, we decided to propose immunosuppressive treatment with topical cyclosporine to avoid long-term treatment with topical corticosteroids. The aim is to strike ichthyosis by maintaining the tear film quality and, simultaneously, to reduce levels of ocular surface inflammation, mitigating acute crises that can endanger a long-term worse prognosis for the conjunctival and corneal tissues throughout the child's life.
This clinical case demonstrates the preponderance that the ocular surface can assume in rare but incapacitating systemic diseases and the importance of a timely diagnosis, stratification, and a step-up treatment approach. This is the key to avoiding long-term complications, which may result in significant loss of life quality and, ultimately, irreversible blindness.
Statements
Ethics approval and consent to participate
Ethical approval is not required for this study by local or national guidelines. The patient's parents (given the patient's age under 18) informed consent was obtained during the assessment visits.
Consent for publication
Written informed consent was obtained from the patient's parents (given the patient's age under 18) for publication of this case report and any accompanying images.
Conflict of Interest Statement: Miguel Mesquita Neves is consultant of Alcon Portugal – Produtos e Equipamentos, Lda.
The other authors have no conflicts of interest to declare.
Funding Sources: Nothing to declare - the research and publication of this article were not funded.
Acknowledgment (optional): Nothing to declare.
Author Contributions
JL combined the case design, data acquisition, and manuscript drafting and finalized the manuscript; PB combined the case design, data acquisition, and manuscript critical review; MN conducted data analysis and manuscript critical review; MG conducted data analysis and critical manuscript review. LO conducted data analysis, critically reviewed the manuscript, performed the medical treatment, and conducted the patient's follow-up. All authors approved the final version of the manuscript and are accountable for all aspects of the work. All authors attest that they meet the current ICMJE criteria for authorship.
Data Availability Statement
The clinical data supporting this clinical case's findings are available in the electronic hospital register of CHUPorto. All data generated or analyzed during this study are included in this article. Further inquiries can be directed to the corresponding author.
References
- Al-Amry MA (2016) Ocular manifestation of Ichthyosis. Saudi J Ophthalmol. 30(1): 39–43.
- Jay B, Blach RK, Wells RS (1968) Ocular manifestations of ichthyosis. Br J Ophthalmol. 52(3): 217–26.
- Wells RS, Kerr CB (1966) Clinical Features of Autosomal Dominant and Sex-linked Ichthyosis in an English Population. Br Med J. 1(5493): 947–50.
- Cinar Y, Selcuk CT, Cingu AK, Turkcu FM, Yuksel H, et al. (2014) Spontaneous bilateral corneal perforation in a patient with ichthyosis. Int Ophthalmol. 34(4): 919–21.
- Vahlquist A, Gånemo A, Virtanen M (2008) Congenital ichthyosis: An overview of current and emerging therapies. Acta Derm Venereol. 88(1): 4–14.
- Zdebik A, Zdebik N, Fischer M (2021) Ocular manifestations of skin diseases with pathological keratinization abnormalities. Postep Dermatologii i Alergol. 38(1): 4–14.
- Malhotra R, Hernández-Martın A, Oji V (2018) Ocular manifestations, complications and management of congenital ichthyoses: a new look. Br J Ophthalmol. 102(5): 586–592.
- Alyasin S, Esmaeilzadeh H, Ebrahimi N, Nabavizadeh SH, Kashef S, et al. (2019) Phenotyping and long-term follow up of patients with hyper IgE syndrome. Allergol Immunopathol (Madr). 47(2): 152–158.
- Minegishi Y (2021) Hyper-IgE syndrome, 2021 update. Allergol Int. 70(4): 407–414.
- Lavoie A, Rottem M, Grodofsky MP, Douglas SD (1989) Anti- Staphylococcus aureus IgE Antibodies for Diagnosis of Hyperimmunoglobulinemia E-Recurrent Infection Syndrome in Infancy. Am J Dis Child. 143(9): 1038–1041.
- Grimbacher B, Holland SM, Gallin JI, Greenberg F, Hill SC, et al. (1999) Hyper-IgE syndrome with recurrent infections--an autosomal dominant multisystem disorder. N Engl J Med. 340(9): 692–702.
- Tsuge I, Morishita M, Kato T, Tsutsumi M, Inagaki H, et al. (2015) Identification of novel FATP4 mutations in a Japanese patient with ichthyosis prematurity syndrome. Hum Genome Var. 2: 15003.