


Seda Yılmaz1*, Özlen Bektaş2, Özcan Çeneli3, Sinan Demircioğlu3, Pembe Oltulu4, Mehmet Alparslan Yılmaz5
1Konya City Hospital, Department of Internal Medicine, Clinic of Hematology, Konya, Turkey
2Karadeniz Technical University, School of Medicine, Department of Haematology, Trabzon, Turkey
3Necmettin Erbakan University, Meram Faculty of Medicine, Department of Hematology, Konya, Turkey
4Necmettin Erbakan University, Meram Faculty of Medicine, Department of Pathology, Konya, Turkey
5Fizikon Medical Center
*Corresponding Author: Seda Yılmaz, Konya City Hospital, Department of Internal Medicine, Clinic of Hematology, Konya, Turkey.
Abstract
Purpose: Kikuchi-Fujimoto's disease (KFD) is a rare, idiopathic, and usually self-limiting condition with an unclear etiology. It is characterized by fever and cervical lymphadenopathy, requiring a lymph node biopsy to establish an exact diagnosis. KFD has rarely been reported in association with systemic lupus erythematosus (SLE), occurring either before, concurrently, or after the onset of KFD.
Methods and Results: Herein, we report a 19-year-old male patient with fever and lymphadenopathy who subsequently developed autoimmune hemolytic anemia and renal failure.
Conclusion: Determining the relationship between KFD and SLE is of clinical importance in initiating early and intensive immunosuppressive therapy. Patients should be followed for relapses or development of SLE for several years.
Key Words: Hemolytic anemia; Kikuchi-Fujimoto's disease; lymphadenopathy; renal failure; systemic lupus erythematosus
Introduction
Kikuchi-Fujimoto's disease (KFD) is a rare, idiopathic, and usually self-limiting condition of an unknown etiology [1]. It is characterized by fever and cervical lymphadenopathy, and the exact diagnosis requires a lymph node biopsy. Despite clinical and histological similarities to systemic lupus erythematosus (SLE), tuberculosis, and lymphoma, KFH should be differentiated from these conditions since the prognosis and management differ dramatically. Also, KFH and SLE may rarely occur in association with each other, with a diagnosis of KFH established before, concurrently, or after a diagnosis of SLE [2,3].
Case Report
A 19-year-old male patient presented to an outpatient facility of an external center with a 2-week history of high fever, rash in the face and trunk, and a painful cervical mass and was subsequently admitted to the internal medicine clinic. He was referred to our unit due to a fever unresponsive to antibiotherapy and anemia. Physical examination showed a body temperature of 39 C, painless lymph nodes (15 to 20 mm) in cervical, axillary, and inguinal areas, and hepatosplenomegaly. Other physical exam findings were normal. Laboratory work-up revealed the following: erythrocyte sedimentation rate 23 mm/sec; lymphopenia (1000/mm3), anemia (hemoglobin 8.8 g/dl), normal creatinine (0.6 mg/dl), increased lactate dehydrogenase (414 U/L; normal: 125-220), increased bilirubin (total bilirubin 2.2 mg/dl, indirect bilirubin 1.6 mg/dl), decreased iron, increased iron binding capacity and ferritin (371 ng/ml; normal: 14-150). Urinalysis was unremarkable. No microbiological growth occurred in blood and urinary cultures. Serology for HIV, CMV, parvovirus B19, Toxoplasma, and Epstein- Barr virus was negative. Peripheral blood smear showed hypochromic erythrocytes with anisocytosis. Echocardiography showed no cardiac pathology. An excisional biopsy of the axillary lymph node showed necrotizing lymphadenitis (Figure 1).
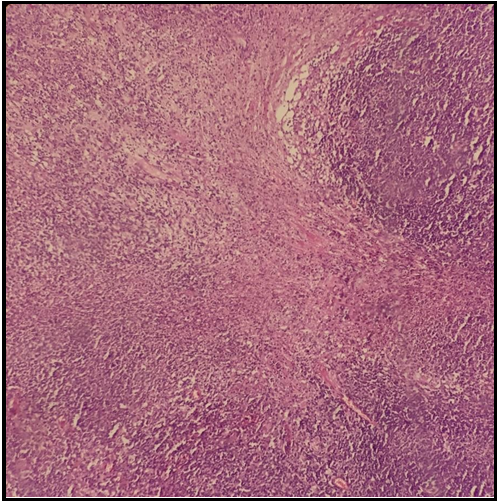
Figure 1. Necrotizing lymphadenitis in excisional biopsy from the axillary lymph node.
A diagnosis of KH was made based on fever, lymphadenopathy, and biopsy-confirmed necrotizing lymphadenitis. During the course of the diagnostic work-up, an increase in creatinine (2.9 mg/dl) and indirect bilirubin and a decrease in haptoglobin (< 8 mg/dl) and hemoglobin (5.4 g/dl) occurred. The Coombs test was positive, and 24-hour urinary protein was 430 mg/day. The antinuclear antibody (ANA) titer was 1/1000, with positivity exhibiting a homogenous and granular pattern. Anti-ds DNA antibody and anti-cardiolipin IgM were positive. C3 (0.6 g/L, normal: 0.9-1.8) and C4 (0.06 g/L, normal: 0.1-0.4) were significantly reduced, while direct and indirect Coombs tests were negative. Also, a diagnosis of SLE was made based on ANA, anti-ds DNA, anti-cardiolipin antibody positivity, hemolytic anemia, lymphopenia, urine findings, elevated creatinine, and reduced complement levels. A renal biopsy was scheduled but not performed due to the patient's refusal. For management, monthly pulse cyclophosphamide and high-dose methyl-prednisolone were commenced due to renal involvement. The treatment resulted in the normalization of creatinine and the elevation of hemoglobin levels.
Discussion
Kikuchi's disease, also known as Kikuchi-Fujimoto disease or Kikuchi histiocytic necrotizing lymphadenitis, is a benign and usually self-limiting type of histiocytic necrotizing lymphadenitis. Patients may present with lymphadenopathy, fever, myalgia, arthralgia, and, more rarely, hepatosplenomegaly. Although it has been associated with SLE, it tends to follow a benign clinical course with spontaneous remission expected to occur within four months.
Its pathogenesis remains unknown. The immune responsivity of the T cells and histiocytes has been proposed to occur in these patients based on its clinical presentation, course, and histological changes. Also, Epstein-Barr virus (EBV), human herpes virus 6, human herpes virus 8, human immune deficiency virus (HIV), parvovirus B19, paramyxoviruses, para-influenza virus, yersinia enterocolitica, and toxoplasma have been implicated [4-9].
Although initially described in female patients, it may also occur in males with a female-to-male ratio of approximately 4 to 1 [10].
In a retrospective study of 244 KFH patients, the most common symptoms included high fever (35 %), fatigue (7 %), and joint pain (7 %) [11], while the most common clinical and laboratory signs were lymphadenopathy (100 %), rash (10 %), arthritis (7 %), hepatosplenomegaly (3 %), leukopenia (43 %), elevated erythrocyte sedimentation rate (40 %), and anemia (23 %). Lymph node enlargement is generally moderate (1 to 2 cm in diameter), while some patients may have larger lymph nodes (≤ 7 cm). Typically, lymph nodes are complex, discrete, mobile, and have a smooth surface [12]. Blunt or acute pain may commonly occur with lymph node enlargement [12]. On the other hand, fever, lymphadenopathy, splenomegaly, and hepatomegaly have been reported to occur in 36 %, 7-16 %, 5 %, and 2 % of patients with SLE, respectively. Our patient had a fever, lymphadenopathy, hepatosplenomegaly, and anemia. The diagnosis of KFH is based on lymph node biopsy showing the presence of histiocytic cellular infiltration. KFH and SLE share specific histological characteristics, and the tubuloreticular structures observed in the lymphocyte, and endothelial cells in SLE patients have some resemblance to those in Kikuchi disease. SLE- specific antibodies, hematoxylin bodies, and vasculitis in lupus lymphadenitis are assistive in the differential diagnosis [13, 14, 15]. In our case, the absence of hematoxylin bodies, as well as the absence of significant vasculitis, pointed away from a diagnosis of SLE lymphadenopathy.
Currently, there are no effective therapeutic strategies for KFH, with a usual resolution of signs and symptoms within 1 to 4 months. Patients should be followed-up for several years for relapses or the development of SLE. Monthly pulse cyclophosphamide plus high-dose methylprednisolone were used to treat our patient after a diagnosis of SLE was made. In the subsequent course of his condition, creatinine levels were normalized, and hemoglobin levels were increased. Identifying the association between KFH and SLE is of clinical significance in initiating early and intensive immunosuppressive therapy.
Conflicts of interest: The authors certify that there is no conflict of interest with any financial organization regarding the material discussed in the manuscript.
Disclaimer: None.
Funding Sources: None.
References
- Gülez, P, Hızarcıoğlu M, Ortaç R (2013) Kikuchi Fujimoto hastalığı ile birlikte hemofagositik sendrom. Türk Pediatri Arşivi. 48(1): 71 – 72.
- Santana A, Lessa B, Galrão L, Lima I, Santiago M (2005) Kikuchi–Fujimoto’s disease associated with systemic lupus erythematosus: Case report and review of the literature. ClinRheumatol. 24(1): 60–3.
- Zuo Y, Foshat M, Qian YW, Kelly B, Harper B, Karnath B, et al. (2012) A rare case of Kikuchi Fujimoto’s disease with subsequent development ofsystemic lupus erythematosus. Case Rep Rheumatol. 2012:325062.
- Yen A, Fearneyhough P, Raimer SS, Hudnall SD (1997) EBV- associated Kikuchi's histiocytic necrotizing lymphadenitis with cutaneous manifestations. J Am AcadDermatol. 36(2 Pt 2): 342- 6.
- Hudnall SD, Chen T, Amr S, Young KH, Henry K (2008) Detection of human herpesvirus DNA in Kikuchi-Fujimoto disease and reactive lymphoid hyperplasia. Int J ClinExpPathol. 1(4): 362-8.
- Yufu Y, Matsumoto M, Miyamura T, Nishimura J, Nawata H, et al. (1997)Parvovirus B19-associated haemophagocytic syndrome with lymphadenopathy resembling histiocytic necrotizing lymphadenitis (Kikuchi's disease). Br J Haematol. 96(4): 868-71.
- Chiu CF, Chow KC, Lin TY, Tsai MH, Shih CM, et al. (2000) Virus infection in patients with histiocytic necrotizing lymphadenitis in Taiwan. Detection of Epstein-Barr virus, type I human T-cell lymphotropic virus, and parvovirus B19. Am J ClinPathol. 113(6): 774-81.
- Hudnall SD (2000) Kikuchi-Fujimoto disease. Is Epstein-Barr virus the culprit? Am J ClinPathol. 113(6): 761-4.
- Huh J, Kang GH, Gong G, Kim SS, Ro JY, et al. (1998) Kaposi's sarcoma-associated herpesvirus in Kikuchi's disease. Hum Pathol. 29(10): 1091-6.
- Meyer O (1999) Kikuchi disease. Ann Med Interne(Paris). 150(3): 199-204.
- Kucukardali Y, Solmazgul E, Kunter E, Oncul O, Yildirim S, et al. (2007) Kikuchi-Fujimoto Disease: analysis of 244 cases. ClinRheumatol 26(1): 50-4.
- Özmen M, Altıner NN, Topçugil F, Ermete M, Aslan SL (2014) Kikuchi hastalığı ve sistemik lupus eritematoz: Olgu sunumu. Turkish Dermatology &Venerology Turkderm. 48(1): 47-50.
- Dorfman RF, Berry GJ (1988) Kikuchi’s histiocytic necrotizing lymphadenitis: an analysis of 108 cases with emphasis on differential diagnosis. Semin DiagnPathol. 5(4): 329-45.
- Martínez-Vázquez C, Hughes G, Bordon J, Alonso-Alonso J, Anibarro-Garcia A, et al. (1997) Histiocytic necrotizing lymphadenitis, Kikuchi-Fujimoto’s disease, associated with systemic lupus erythematosus. QJM. 90(8): 531-3.
- Menasce LP, Banerjee SS, Edmondson D, Harris M (1998) Histiocytic necrotizing lymphadenitis (Kikuchi-Fujimoto disease): continuing diagnostic difficulties. Histopathology. 33(3): 248-54.