


Abebe Habtamu Tamire1*, Tenagne Million2
1Department of Pediatrics and child health, Addis Ababa University, College of Health Sciences, Addis Ababa, Ethiopia.
2Out Patient Department, Meron medium clinic, Addis Ababa, Ethiopia.
*Corresponding Author: Abebe Habtamu Tamire, Department of Pediatrics and child health, Addis Ababa University, College of Health Sciences, Addis Ababa, Ethiopia.
Abstract
Nieman-Pick disease, also known as sphingomyelin cholesterol lipidosis, is a rare storage disease primarily affecting children. It is classified into three types: A, B, and C. Types A and B are caused by mutations in the sphingomyelin phosphodiesterase 1 gene, leading to a deficiency in acid sphingomyelinase activity. This enzymatic deficiency results in the pathological accumulation of sphingomyelin in the monocyte- macrophage system. In Type A, the progressive deposition of sphingomyelin in the central nervous system leads to a neurodegenerative course, while in Type B, non-neuronal tissues are affected, resulting in systemic disease manifestations.
Type C, on the other hand, is caused by mutations in the NPC-1 and NPC-2 genes, which impair the cellular processing and transport of low- density lipoprotein. This disruption in lipid metabolism contributes to the development of Niemann-Pick disease Type C.
Clinical features commonly associated with Niemann-Pick disease include hepatosplenomegaly (enlargement of the liver and spleen), neurodegenerative symptoms, and interstitial lung disease. These symptoms can significantly impact the quality of life and overall prognosis of affected individuals, particularly children.
This case report focuses on Niemann-Pick disease in Ethiopia, highlighting the importance of recognizing and managing this rare disorder in the pediatric population.
Keywords: Niemann-Pick disease, Hepatosplenomegaly, Neurodegenerative, Interstitial lung disease, Children, Ethiopia.
What is new/known: This is a rare case in Ethiopia and helps as a teaching aid.
Introduction
Nieman-Pick disease, also known as sphingomyelin cholesterol lipidosis, is a rare storage disease primarily affecting children. It is subdivided into three types: A, B, and C. Type A and B are caused by mutations in the sphingomyelin phosphodiesterase 1 gene, resulting in a primary deficiency of acid sphingomyelinase activity [1,2].
Type A (NPD-A) is the acute neuronopathic form of the disease and is more prevalent among Ashkenazi Jews. The estimated prevalence of Type A and B is 1 in 250,000 individuals. Infants with NPD-A appear normal at birth but experience failure to thrive, hepatosplenomegaly (enlargement of the liver and spleen), moderate lymphadenopathy, and psychomotor retardation by 6 months of age. The storage of sphingomyelin in pulmonary macrophages leads to interstitial lung disease, recurrent infections, respiratory failure, and a rapid and profound loss of neurologic function, ultimately resulting in death between 2 and 3 years of age [3,4].
Niemann-Pick Disease Type B (NPD-B) affects individuals from various ethnic backgrounds and has a later onset and less severity compared to Type A. Most cases are diagnosed in infancy and childhood when liver and spleen enlargement is observed.
Thrombocytopenia due to hypersplenism is common among affected patients. Additional manifestations include short stature with delayed skeletal maturation, interstitial lung disease, hyperlipidemia, and ocular abnormalities. The natural history of NPD-B involves progressive hypersplenism and gradual deterioration of pulmonary function. Patients typically exhibit mild pulmonary involvement during diagnosis, and neurologic abnormalities are absent in most cases [5-7].
Niemann-Pick Disease Type C (NPD-C) displays highly variable age of onset and clinical features, with less severe hepatosplenomegaly compared to Types A and B. More than 85% of patients with NPD-C exhibit systemic involvement of the liver, spleen, or lung. The onset of the disease usually occurs in the middle to late childhood, following a period of normal early development. Cerebellar involvement characterized by clumsiness and gait problems is joint in NPD-C and progresses to frank ataxia and slow cognitive deterioration. Progressive dystonia and dysphagia may impair oral feeding, and death often results from aspiration pneumonia during the second or third decade of life [8-10].
In summary, Niemann-Pick disease is a rare storage disease that affects children and is classified into three types. Type A and B present with primary deficiency of acid sphingomyelinase activity, leading to severe neurologic and systemic manifestations, while Type C exhibits variable clinical features and impacts the liver, spleen, lung, and neurologic function. Understanding the different types and their characteristics is crucial for accurately diagnosing and managing Niemann-Pick disease.
Diagnosis
Diagnosis of Niemann-Pick disease involves a combination of laboratory tests, histology, electron microscopy, fundoscopy, and enzyme assays. The following diagnostic methods are commonly used:
1. Laboratory Tests:
1. Low levels of high-density lipoprotein (HDL)
2. High levels of low-density lipoprotein (LDL)
3. Hypertriglyceridemia (elevated triglyceride levels)
2. Histology:
Examination of tissue samples reveals the presence of sizeable lipid- laden foam cells in various locations, including the reticuloendothelial cells of the spleen, bone marrow, blood vessels, central nervous system (CNS), and retinal cells.
3. Electron Microscopy:
The use of electron microscopy allows for the visualization of specific cellular features, such as lysosomal and myelin inclusions, which are characteristic of Niemann-Pick disease.
4. Fundoscopy:
Fundoscopy, or examination of the retina, may reveal the presence of macular cherry red spots. This finding is typically observed in Type A and Type B Niemann-Pick disease.
5. Enzyme Assay:
Definitive diagnosis of Type A and Type B Niemann-Pick disease requires the demonstration of sphingomyelinase activity that is less than 10% of controls. Enzyme assays measure the activity level of specific enzymes involved in lipid metabolism.
6. Filipin Staining:
For the definitive diagnosis of Niemann-Pick disease Type C, abnormal intracellular cholesterol accumulation is demonstrated in cultured fibroblasts using filipin staining. This staining technique allows for the visualization of characteristic cholesterol-filled vesicles or inclusions.
By employing these diagnostic methods, healthcare professionals can accurately identify and classify the different types of Niemann-Pick disease based on the specific laboratory, histological, and microscopic findings.
Case Report
Patient Information:
Age: 9 years old
Gender: Male
Clinical History:
1. The patient was relatively healthy until age 3 when abdominal swelling began to develop. Initially small, the swelling gradually increased in size over time.
2. The family attributed the swelling to weight gain and did not seek medical care initially.
3. 1 year and 2 months before the current presentation, the patient experienced increased abdominal swelling, significant but unquantified weight loss, drenching night sweats, dry intermittent cough (non-whooping, non-barking), and loss of appetite.
4. The patient visited a nearby hospital, where a normal chest X-ray (CXR) and regular GeneXpert test from sputum were reported.
5. Despite treatment for pneumonia, including a 6-month course of anti-TB medication, there was no improvement in symptoms, and the swelling remained the same.
6. The patient was then started on hydration and allopurinol due to consideration of hepatosplenomegaly (HSM) secondary to possible Non-Hodgkin's Lymphoma (NHL).
7. However, after further evaluation, with no clinical or biochemical evidence for NHL, the medications were discontinued.
8. The patient presented to the current hospital with worsening abdominal swelling of 2 weeks' duration, along with a dry intermittent cough, shortness of breath, and easy fatigability.
9. No other complaints were reported.
Physical Examination
General appearance: The patient appears acutely sick and is in respiratory distress.
Vital signs:
1. Blood pressure: 100/64 mmHg
2. Pulse rate: 96 beats/min, regular and complete in volume
3. Respiratory rate: 72 breaths/min
4. Temperature: 36.2°C (axillary)
5. Oxygen saturation: 88% on atmospheric air
6. Random blood sugar: 92 mg/dL
Anthropometry:
1. Weight: 24 kg
2. Height: 109 cm
3. Mid-upper arm circumference (MUAC): 15 cm
4. Body mass index (BMI): 20.2 kg/m²
5. Body mass index for age: Between 1-2 z-scores
6. Height for age: Less than -3 z-scores (severe short stature)
Physical examination findings:
1. Pale conjunctivae
2. Subcostal-intercostal retractions
3. Bilateral basal fine crepitations on lung auscultation
4. Grossly distended abdomen with no evidence of ascites
5. Palpable liver 11 cm below the right costal margin, with a total vertical liver span (TVLS) of 16 cm
6. Palpable spleen measuring 20 cm
7. Some palmar pallor
8. No other physical findings were observed.
Based on the provided history and physical examination findings, the patient presents with a significant clinical concern, including severe short stature, hepatosplenomegaly, respiratory distress, and anemia. Further investigations and management are warranted to determine the underlying cause and provide appropriate treatment.
Investigations:
1. Urinalysis (U/A): Non-revealing.
2. Serum electrolytes: Normal.
3. Serological tests:
Hepatitis B surface antigen (HBsAg): Negative.
Hepatitis C virus (HCV) antibody: Negative.
HIV screening: Negative.
1. Liver function tests (LFT) and renal function tests (RFT): Normal.
2. Antinuclear antibody (ANA): Negative.
3. Serum albumin levels: Initial level of 2.5 mg/dL, increased to 3.78 mg/dL after albumin transfusion.
4. GeneXpert test from sputum: Negative.
5. Chest X-ray (CXR): Bilateral infiltrates suggestive of interstitial lung disease.
6. Abdominal ultrasound (U/S):
Hepatomegaly with a size of 14.5 cm.
Huge splenomegaly with focal wedge-shaped infarction measuring 20 cm.
7. Peripheral Blood Morphology Table 1:
Red blood cells (RBC): Normocytic-normochromic.
White blood cells (WBC): Adequate with 60% neutrophils, 36% lymphocytes, 1% monocytes, and 3% eosinophils.
8. Peripheral morphology and bone marrow aspiration (PM+BMA): Erythroid hyperplasia observed in both peripheral morphology and bone marrow aspiration.
9. CT scan of the chest Confirms the presence of interstitial lung disease.
10. Bone marrow histology: Shows large lipid-laden foam cells, characteristic of Niemann-Pick disease type C (NPD-C) Figure 1.
11. Liver histology: Demonstrates sheets of round to polygonal multivacuolated foam cells with abundant cytoplasm and central round nuclei, along with average liver parenchyma Figure 2.
12. Histologic sections of bone marrow: Show hematopoietic marrow elements and areas of sheets of round to polygonal macrophages with vacuolated cytoplasm and central round nuclei Figure 3.
Based on the investigations, the patient's condition is characterized by hepatosplenomegaly, interstitial lung disease, erythroid hyperplasia in the bone marrow, and the presence of large lipid-laden foam cells in various tissues. These findings are consistent with a diagnosis of Niemann-Pick disease type C (NPD-C). Further evaluation and management should be focused on confirming the diagnosis and providing appropriate treatment for NPD-C.
Table 1: shows weekly CBC result of the patient
|
CBC |
19/06/23 |
26/06/23 |
03/07/23 |
12/07/23 |
19/07/23 |
|
WBC |
4880 |
4340 |
3360 |
1700 |
2100 |
|
N% |
48.9 |
37.8 |
37.5 |
6 |
10.2 |
|
L% |
41.9 |
52.9 |
50.8 |
73.4 |
63.3 |
|
Hbg |
9.8 |
8.4 |
9 |
8.6 |
9.8 |
|
ANC |
2390 |
1640 |
1370 |
200 |
400 |
|
HCT |
30 |
26.4 |
26.1 |
25.5 |
28.4 |
|
PLT |
178,000 |
115,000 |
92,000 |
164,000 |
162,000 |
|
MCV |
75.4 |
76.4 |
80.6 |
77.2 |
75.7 |
|
RDW |
18.8 |
19.1 |
19.5 |
21.2 |
19.5 |
|
Eosinophil |
2.3 |
1.6 |
2.2 |
2.3 |
2.1 |
|
ESR |
100mm/hr |
|
65mm/hr |
|
|
|
CRP |
negative |
negative |
|
|
|
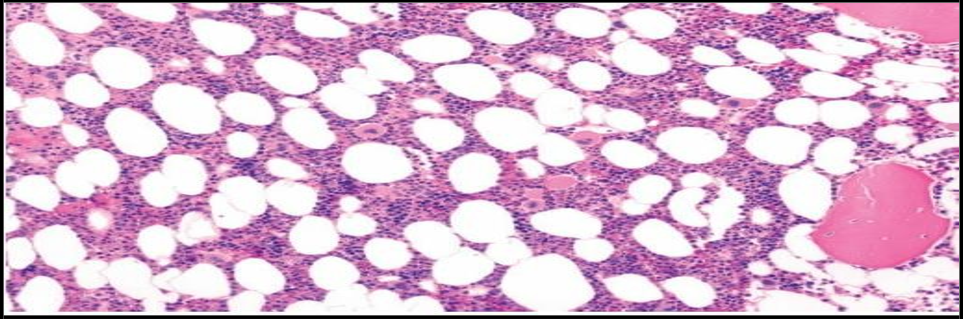
Figure 1: Bone barrow histology showing large lipid laden foam cells (Characteristics of NPD-C) in our patient.
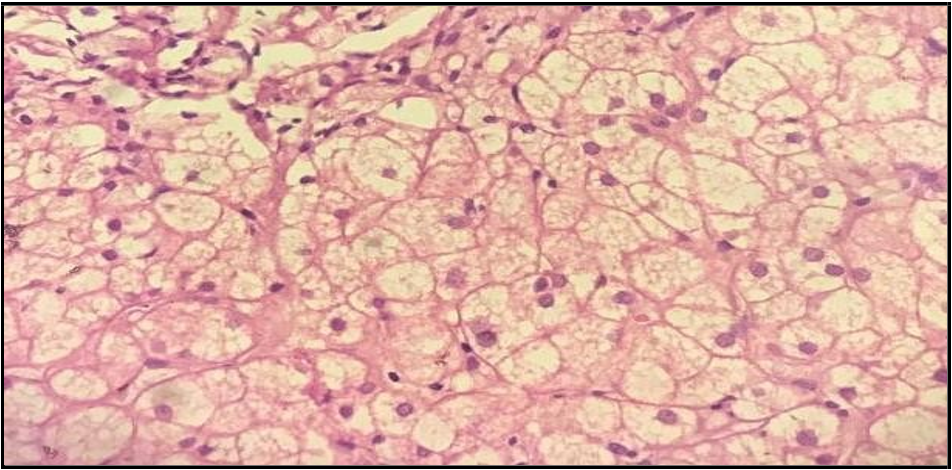
Figure 2: Histologic section from the liver of our patient shows mainly sheets of round to polygonal multivacuolated foam cells having abundant multivacuolated cytoplasm and central round nuclei, with normal liver parenchyma
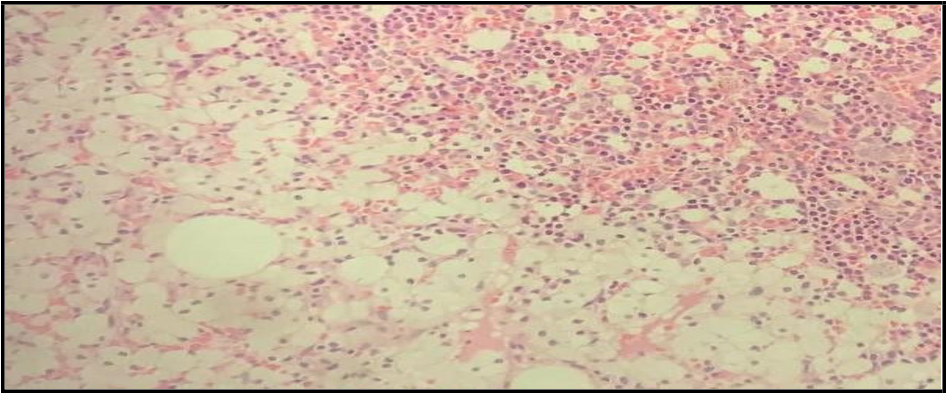
Figure 3: Histologic sections show hematopoietic marrow elements and areas of sheets of round to polygonal macrophages having vacuolated cytoplasm and central round nuclei.
Discussion
The presented case report suggests a diagnosis of Niemann-Pick disease type C (NPD-C) based on the clinical features, laboratory investigations, and histological findings. NPD-C is a rare autosomal recessive lysosomal storage disorder characterized by defective intracellular lipid trafficking and the accumulation of cholesterol and glycosphingolipids in various tissues [11].
The age of onset and clinical manifestations of NPD-C can vary widely. In this case, the patient's symptoms began around age 3, with the development of abdominal swelling. The subsequent progression of symptoms, including weight loss, drenching night sweats, dry intermittent cough, and loss of appetite, raised concerns for an underlying systemic disease. The initial evaluation, including CXR, gene Xpert test, and anti-TB treatment, did not provide improvement, leading to further investigations.
The physical examination revealed hepatosplenomegaly, respiratory distress, and anemia. Laboratory tests showed normal serum electrolytes, negative viral hepatitis and HIV screening, normal LFT and RFT, and a low serum albumin level [12]. The CXR and CT scan confirmed the presence of interstitial lung disease. Abdominal ultrasound demonstrated hepatomegaly and splenomegaly with a focal wedge-shaped infarction. Bone marrow aspiration and peripheral blood morphology revealed erythroid.
The definitive diagnosis of NPD-C is established by demonstrating abnormal intracellular cholesterol accumulation in cultured fibroblasts using filipin staining [5,8]. However, in this case report, the diagnosis of NPD-C was established based on the supportive clinical features, laboratory investigations, and histological findings, which strongly suggested the presence of lipid storage disease.
It is worth noting that the diagnosis of NPD-C in this case report represents the first reported case of a storage disease being diagnosed in Ethiopia. This highlights the importance of considering rare genetic disorders in the differential diagnosis, even in settings with limited resources and less familiarity with these conditions.
Conclusion
Based on the presence of hepatosplenomegaly, findings from the CXR, CT scan of the abdomen, liver, and bone marrow histologies, the overall evidence suggests a diagnosis of Niemann-Pick disease. It is worth noting that this is the first reported case of a storage disease being diagnosed in Ethiopia. Further investigations and specialized testing, such as filipin staining to confirm abnormal intracellular cholesterol accumulation, may be warranted to establish a definitive diagnosis of Niemann-Pick disease type C (NPD-C).
Conflict of interest: No Conflict of interest.
Consent: Informed consent has been duly obtained from the patient's legal guardian, and assent has been obtained from the patient, allowing for the publication of this case report and any accompanying images.
References
- Thurm A, Chlebowski C, Joseph L, Farmer C, Adedipe D, et al. (2020) Neurodevelopmental Characterization of Young Children Diagnosed with Niemann-Pick Disease, Type C1. J Dev Behav Pediatr. 41(5): 388-396.
- Eskes ECB, Sjouke B, Vaz FM, Goorden SMI, van Kuilenburg ABP, et al. (2020) Biochemical and imaging parameters in acid sphingomyelinase deficiency: Potential utility as biomarkers. Mol Genet Metab. 130(1): 16-26.
- Xu Y, Zhang Q, Tan L, Xie X, Zhao Y (2019) The characteristics and biological significance of NPC2: Mutation and disease. Mutat Res Rev Mutat Res. 782: 108284.
- Bianconi SE, Hammond DI, Farhat NY, Dang Do A, Jenkins K, et al. (2019) Evaluation of age of death in Niemann-Pick disease, type C: Utility of disease support group websites to understand natural history. Mol Genet Metab. 126(4): 466-469.
- Kresojević N, Mandić-Stojmenović G, Dobričić V, Petrović I, Brajković L, et al. (2020) Very Late-Onset Niemann Pick Type C Disease: Example of Progressive Supranuclear Palsy Look-Alike Disorder. Mov Disord Clin Pract. 7(2): 211-214.
- Chugani HT (2019) Positron Emission Tomography in Pediatric Neurodegenerative Disorders. Pediatr Neurol. 100: 12-25.
- Jezela-Stanek A, Chorostowska-Wynimko J, Tylki-Szymańska A (2020) Pulmonary involvement in selected lysosomal storage diseases and the impact of enzyme replacement therapy: A state- of-the art review. Clin Respir J. 14(5): 422-429.
- Pará C, Bose P, Pshezhetsky AV (2020) Neuropathophysiology of Lysosomal Storage Diseases: Synaptic Dysfunction as a Starting Point for Disease Progression. J Clin Med. 9(3): 616.
- Cawley NX, Sojka C, Cougnoux A, Lyons AT, Nicoli ER, et al. (2020) Abnormal LAMP1 glycosylation may play a role in Niemann-Pick disease, type C pathology. PLoS One. 15(1): e0227829.
- Patterson M. Niemann-Pick Disease Type C. (2000) In: Adam MP, Mirzaa GM, Pagon RA, Wallace SE, Bean LJH, Gripp KW, Amemiya A, editors. GeneReviews® [Internet]. University of Washington, Seattle; Seattle (WA).
- Breiden B, Sandhoff K (2019) Lysosomal Glycosphingolipid Storage Diseases. Annu Rev Biochem. 88: 461-485.
- Chen KJ, Jin RM, Shi CC, Ge RL, Hu L, et al. (2018) The prognostic value of Niemann-Pick C1-like protein 1 and Niemann-Pick disease type C2 in hepatocellular carcinoma. J Cancer. 9(3): 556-563.